自动确定单细胞聚类数量!?这才是做单细胞分析最需要的 R 包(recall)
生信碱移Recall 通过人工生成随机的噪声假基因判断细胞聚类簇是否拆得过细,如果过细就自动往回合并或者调整分辨率,从而得到优化的聚类。单细胞 RNA 测序(scRNA-seq)能够分析包含数千至数百万个单细胞转录组图谱的数据集。主流分析流程 seurat 与 scanpy 一般包括以下三个大步骤:预处理(质控、归一化、选 HVGs、PCA)无监督聚类(在 PCA 空间构建的邻接图上 执行Louv
生信碱移
recall: 细胞自动聚类
Recall 通过人工生成随机的噪声假基因判断细胞聚类簇是否拆得过细,如果过细就自动往回合并或者调整分辨率,从而得到优化的聚类。
单细胞 RNA 测序(scRNA-seq)能够分析包含数千至数百万个单细胞转录组图谱的数据集。主流分析流程 seurat 与 scanpy 一般包括以下三个大步骤:
-
预处理(质控、归一化、选 HVGs、PCA)
-
无监督聚类(在 PCA 空间构建的邻接图上 执行Louvain或Leiden聚类算法)
-
细胞marker识别,在得到的聚类簇上做差异表达(DE)分析,从而定义细胞类型和marker gene。
经常分析的铁子都知道,单细胞流程分辨率参数、邻居数等都很主观,细胞聚类本身本身是一拍脑袋的操作。换句话说,分辨率调的越大,理论上单细胞簇能够被无限细分。
具体到分析的时候,如果聚类的时候过度细分了,很多 cluster 其实没什么太大差别,注释的时候就麻烦起来了。不仅如此,考虑到单细胞的 FindAllmarker 函数本质上是一对多的差异分析,这个时候无论如何其实都容易检测出差异基因。画热图的时候可能就会发现问题,比如一些聚类具有类似的 marker 基因。
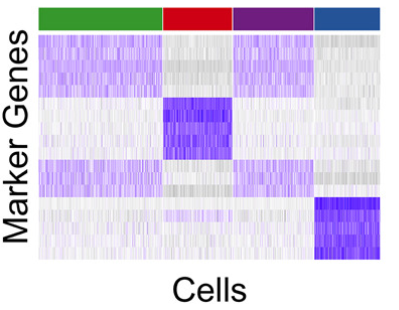
图:聚类簇具有一样的top基因。
前几天看到一个有意思的 R 包 recall,原理比较易懂,代码也非常简单,甚至名称也很好记,这种好东西还是得给各位佬哥佬姐分享分享。R 包对应的文章今年4月3日发表于老牌期刊 AJHG [IF:8.1]。

DOI: 10.1016/j.ajhg.2025.02.014.
简单来说,recall 往基因表达矩阵里放入一批人造假基因,这些基因被设计成不可能代表真实细胞类型差异的阴性对照。然后,recall 对假基因跟真基因一起进行聚类和差异分析。在此基础上,根据假基因与真实基因的结果判断聚类簇是否拆得过细,如果过细就自动往回合并或者调整分辨率,从而得到优化的聚类。
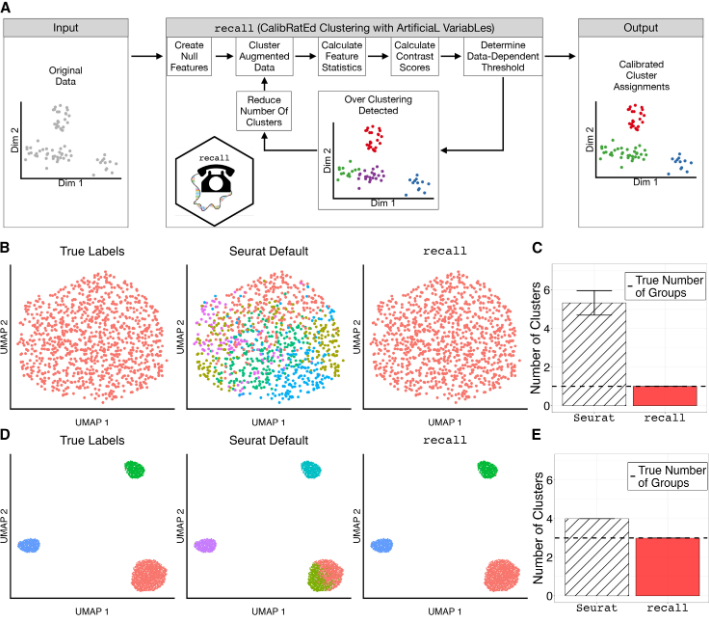
图:recall 方法流程。a.recall 在原始表达矩阵中为每个真实基因构造若干人造基因(作者称为人工变量),使其在边际分布与共变结构上逼近真实基因,但在设计上与细胞类型独立,生物学上仅代表背景噪音。人工基因与真实基因一同参与降维、聚类和差异表达分析。作为负对照:真实的细胞类型差异不应主要体现在这些人工变量上。因此,若任意两个聚类簇之间大量人工基因被判为显著差异基因,则说明当前簇划分缺乏可信的表达差异支撑;此时算法据此合并相关簇或下调聚类分辨率并重新聚类,从而抑制过度聚类并获得统计上更稳健的簇结构。b-d.recall 能够聚类出真实的细胞差异群体。
本文简要介绍 recall 方法的使用流程,感兴趣的铁子可以阅读原文了解过更多信息。
-
https://github.com/lcrawlab/recall
0.R包安装
可以使用如下代码安装该R包:
devtools::install_github("lcrawlab/recall")
# presto并不是一个明确的依赖项,但安装 presto 会使 recall 更快
#devtools::install_github("immunogenomics/presto")
需要注意的是recall仅支持 Seurat v5 版本,如果大家用的是 v4 的话,需要再更新或者同时安装一个 v5 版本的包。这里小编同时安装了v5和v4,所以指定v5的安装路径进行引用:
library(Seurat, lib.loc = "D:/Software/R/R-4.4.0/seuratv5/")
library(SeuratData, lib.loc = "D:/Software/R/R-4.4.0/seuratv5/")
library(recall)
01.recall使用
recall的代码流程非常简单,只需使用FindClustersRecall代替FindClusters函数即可,这里使用10X的外周血单个核细胞数据进行展示。
① 先加载一下数据为 seurat 对象af:
set.seed(123)
af <- readRDS("af.rds")
af <- UpdateSeuratObject(af) # v4结构转v5
② 走一下常规标准化PCA流程:
af <- NormalizeData(af)
af <- FindVariableFeatures(af)
af <- ScaleData(af)
af <- RunPCA(af)
af <- RunUMAP(af, dims = 1:10)
③ 这里展示一下 seurat 流程与 recall 的差异。
首先是seurat的流程,分辨率按照默认选个0.8看看:
# seurat
af_seurat <- FindNeighbors(af, dims = 1:10)
af_seurat <- FindClusters(af_seurat, resolution = 0.8)
然后是recall的流程,直接使用FindClustersRecall函数替换即可:
# recall
af_recall <- FindClustersRecall(af,
resolution_start = 0.8,
reduction_percentage = 0.2,
num_clusters_start = 20,
dims = 1:10, # KNN使用的PCA维度
algorithm = "louvain")
几个参数如下,具体的细节可以再看看原文:
-
resolution_start = 0.8:设置初始聚类分辨率,数值越大初始簇越多,作为起点供后续合并校准。
-
reduction_percentage = 0.2:每次发现过度聚类时收缩聚类复杂度的比例(20%),控制逐步合并/降分辨率的程度。
-
num_clusters_start = 20:指定初始期望簇数规模,大致从约 20 个簇起步再由算法自动调整。
-
dims = 1:10:用于构建 KNN 图和聚类的 PCA 维度范围,这里使用前 10 个主成分。
-
algorithm = "louvain":选择使用 Louvain 图社区检测算法进行聚类。
最后比较一下两者的结果:
p1 <- DimPlot(af_seurat) + ggplot2::ggtitle(label = "Seurat cluter")
p2 <- DimPlot(af_recall) + ggplot2::ggtitle(label = "Recall cluter")
p1+p2
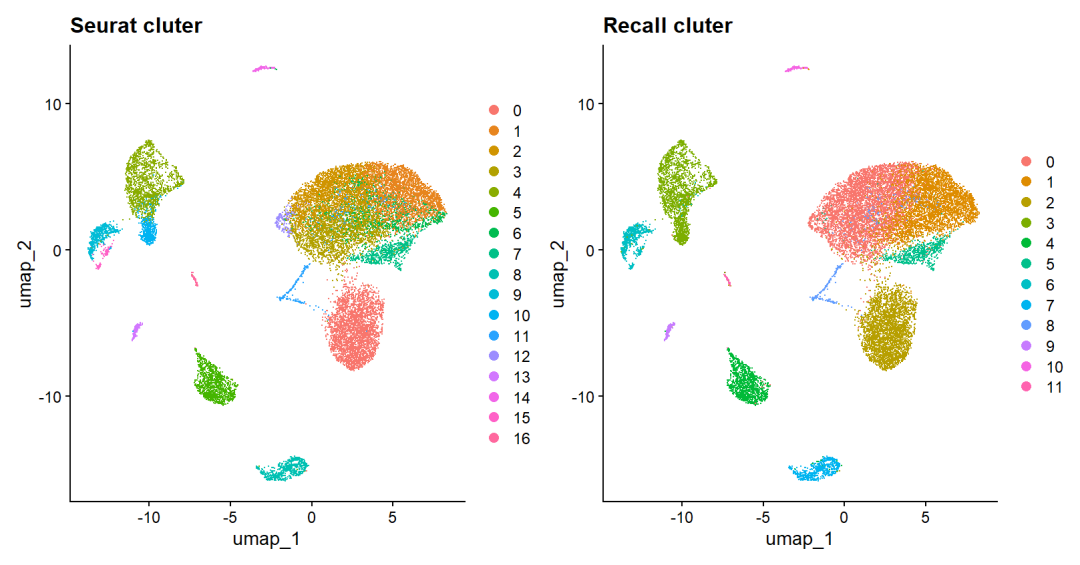
高下立判
这不得赶紧试试

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐


 15
15 0
0- 0



 已为社区贡献10条内容
已为社区贡献10条内容

所有评论(0)