表观基因组分析实操指南:从实验设计到多组学整合
表观基因组分析已从群体细胞水平迈向单细胞、长读段时代,scNanoATAC-seq2 等新技术的出现,为解析胚胎发育、疾病异质性等复杂生物学问题提供了强大工具。实操中需牢记 “实验设计为基础,数据质控为核心,多组学整合为方向” 的原则,灵活选择技术方案。未来,随着 AI 辅助的调控网络重构、空间表观组学技术的发展,表观基因组学将在精准医疗、再生医学等领域发挥更大作用。
引言:解码基因调控的 “隐形开关”
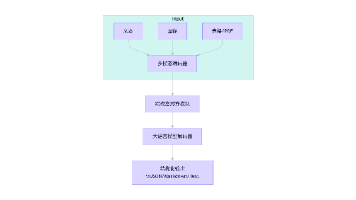
传统遗传学难以解释同卵双胞胎的表型差异、环境诱导的性状遗传等现象 —— 这正是表观遗传学的研究范畴。表观基因组通过 DNA 甲基化、组蛋白修饰、染色质构象等不改变 DNA 序列的调控机制,动态调控基因表达,在发育、衰老、疾病发生中扮演关键角色。随着高通量测序技术的发展,WGBS、ATAC-seq、CUT&Tag 等技术已成为解析表观遗传调控的核心工具。本文将从实验设计、数据处理、高级分析到多组学整合,提供一套可落地的表观基因组分析实操方案。
一、核心技术选择与实验设计(实操基础)
1.1 技术选型决策树
| 研究目标 | 推荐技术 | 关键优势 | 样本要求 |
|---|---|---|---|
| 全基因组 DNA 甲基化(单碱基分辨率) | WGBS | 金标准,覆盖所有 CpG 位点 | 细胞≥1×10⁶,DNA≥1μg |
| 组蛋白修饰 / 转录因子结合位点 | CUT&Tag | 操作简便、噪音低、样本量少 | 细胞≥5×10³,无需甲醛交联 |
| ChIP-seq | 经典方法,抗体选择丰富 | 细胞≥1×10⁶,需甲醛交联 | |
| 染色质可及性(调控元件定位) | ATAC-seq | 快速高效,可单细胞应用 | 细胞≥5×10³,新鲜样本最佳 |
| RNA 修饰(m⁶A 等) | MeRIP-seq | 高特异性富集修饰 RNA | RNA≥10μg,完整性 RIN≥7 |
| RNA - 蛋白相互作用 | RIP-seq | 直接鉴定结合 RNA | 细胞≥1×10⁷,抗体特异性关键 |
1.2 实验设计关键要点
(1)样本制备核心原则
- 细胞样本:新鲜分离后立即处理,避免反复冻融;CUT&Tag 需保持细胞膜完整性,推荐使用 0.05% Digitonin 透化。
- 组织样本:液氮速冻后研磨,ATAC-seq 需先制备细胞核悬液,避免细胞质污染。
- 质量控制:DNA 样本 A260/A280=1.8~2.0,RNA 样本 RIN≥7;ATAC-seq 需检测核小体片段分布(NFR<100bp,单核小体≈200bp)。
(2)对照组设计
- 必设输入对照(Input):用于排除非特异性结合(ChIP-seq/CUT&Tag)。
- 技术重复≥2 次,生物学重复≥3 次;差异分析需满足组内相关系数 > 0.8。
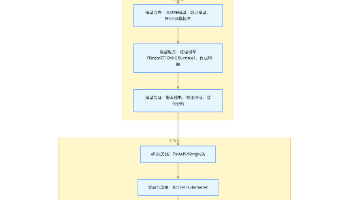
二、通用数据处理流程(代码实操)
2.1 数据预处理(质控→比对→去重)
(1)质量控制与过滤
bash
# 1. FastQC检测原始数据质量
fastqc raw_data/*.fastq.gz -o qc_reports/
# 2. Trim Galore去除接头和低质量碱基(ILLUMINA平台)
trim_galore --illumina --paired --clip_R1 10 --clip_R2 10 \
raw_data/sample1_R1.fastq.gz raw_data/sample1_R2.fastq.gz \
-o clean_data/
关键指标:过滤后 reads 质量 Q30≥85%,接头残留率 < 0.1%。
(2)序列比对
bash
# 1. 构建参考基因组索引(以hg38为例)
bwa index -a bwtsw reference/hg38.fa
# 2. BWA-MEM比对(ATAC-seq/ChIP-seq)
bwa mem -t 8 -M reference/hg38.fa \
clean_data/sample1_R1_trimmed.fq.gz \
clean_data/sample1_R2_trimmed.fq.gz | \
samtools view -Sb -q 30 -o aligned_data/sample1.bam
# 3. 排序与索引
samtools sort aligned_data/sample1.bam -o aligned_data/sample1_sorted.bam
samtools index aligned_data/sample1_sorted.bam
比对后质控:
- 独特映射率≥80%(ATAC-seq 需排除线粒体 DNA,比例 < 10%)
- 用 ATACseqQC 包评估 TSS 富集分数(>5 为合格):
r
library(ATACseqQC)
bamFile <- "aligned_data/sample1_sorted.bam"
tssEnrichment(bamFile, txs = TxDb.Hsapiens.UCSC.hg38.knownGene,
outPlot = "tss_enrichment.pdf")
(3)去重与黑名单过滤
bash
# 1. Picard去除PCR重复
picard MarkDuplicates I=aligned_data/sample1_sorted.bam \
O=aligned_data/sample1_dedup.bam M=duplicate_metrics.txt REMOVE_DUPLICATES=true
# 2. 过滤ENCODE黑名单区域(hg38)
bedtools intersect -v -a aligned_data/sample1_dedup.bam \
-b reference/ENCODE_blacklist_hg38.bed > aligned_data/sample1_final.bam
2.2 核心分析模块(分技术详解)
模块 1:WGBS 数据分析(DNA 甲基化)
(1)甲基化位点 Calling
bash
# Bismark比对(亚硫酸氢盐转化后数据)
bismark --genome reference/hg38/ -1 clean_data/wgbs_sample1_R1.fq.gz \
-2 clean_data/wgbs_sample1_R2.fq.gz -o bismark_results/
# 提取甲基化位点
bismark_methylation_extractor --bedGraph --counts \
bismark_results/sample1_bismark_bt2_pe.bam -o methylation_results/
(2)差异甲基化分析
r
library(methylKit)
# 读取bedGraph文件
methData <- methRead(location = list("sample1_methylation.bedGraph",
"sample2_methylation.bedGraph"),
sample.id = c("control", "treatment"),
assembly = "hg38", context = "CpG")
# 过滤低覆盖度位点(≥10×)
filtered <- filterByCoverage(methData, lo.count = 10, lo.perc = NULL)
# 差异甲基化区域(DMR)识别
dmr <- calculateDiffMeth(filtered, overdispersion = "MN",
adjusted.pvalue.cutoff = 0.05, difference = 25)
# 输出DMR结果
write.table(dmr, "dmr_results.txt", sep = "\t", quote = FALSE)
模块 2:ATAC-seq 数据分析(染色质可及性)
(1)峰识别
bash
# MACS2峰识别(默认参数)
macs2 callpeak -t aligned_data/sample1_final.bam -f BAMPE \
-g hs -n sample1_peaks --outdir peak_results/ --keep-dup all
# 可选:HMMRATAC(ATAC-seq专用,提供核小体定位)
HMMRATAC -b aligned_data/sample1_final.bam -g reference/hg38.fa \
-o hmmratac_results/ -n sample1
峰质量评估:峰数量≥10,000,平均富集倍数≥4,FDR<0.01。
(2)高级分析
- Motif 富集(HOMER):
bash
findMotifsGenome.pl peak_results/sample1_peaks.narrowPeak hg38 \
motif_results/ -size 200 -mask
- 转录因子足迹分析:
r
library(GenomicRanges)
library(footprintR)
bam <- "aligned_data/sample1_final.bam"
peaks <- import.bed("peak_results/sample1_peaks.narrowPeak")
# 偏移校正(+4bp/-5bp)
footprints <- getFootprints(bam, peaks, genome = "hg38")
plotFootprint(footprints, motif = "JASPAR2024_MA0139.1_JUNB")
模块 3:CUT&Tag 数据分析(组蛋白修饰)
(1)峰识别与差异分析
bash
# MACS2峰识别(组蛋白修饰用--broad参数)
macs2 callpeak -t aligned_data/cuttag_sample1.bam -f BAM \
-g hs -n sample1 --outdir cuttag_peaks/ --broad --broad-cutoff 0.1
# DiffBind差异峰分析(R脚本)
library(DiffBind)
sampleSheet <- data.frame(SampleID = c("ctrl1", "ctrl2", "treat1", "treat2"),
Condition = c("Control", "Control", "Treatment", "Treatment"),
bamReads = c("ctrl1.bam", "ctrl2.bam", "treat1.bam", "treat2.bam"),
Peaks = c("ctrl1_peaks.broadPeak", "ctrl2_peaks.broadPeak",
"treat1_peaks.broadPeak", "treat2_peaks.broadPeak"))
dbObj <- dba(sampleSheet = sampleSheet, format = "bam")
dbObj <- dba.count(dbObj, bParallel = TRUE)
dbDiff <- dba.contrast(dbObj, categories = DBA_CONDITION)
dbDiff <- dba.analyze(dbDiff, method = DBA_DESEQ2)
dba.report(dbDiff, file = "diff_peaks.txt")
三、多组学整合分析(进阶实战)
3.1 表观组与转录组整合
(1)流程框架
- ATAC-seq 峰注释到基因(ChIPseeker):
r
library(ChIPseeker)
library(TxDb.Hsapiens.UCSC.hg38.knownGene)
peaks <- readPeakFile("peak_results/sample1_peaks.narrowPeak")
anno <- annotatePeak(peaks, tssRegion = c(-3000, 3000),
TxDb = TxDb.Hsapiens.UCSC.hg38.knownGene)
- 关联差异表达基因(RNA-seq):
r
# 读取RNA-seq差异基因
degs <- read.table("rna_seq_degs.txt", header = TRUE)
# 交集分析
peak_genes <- unique(anno$geneId)
overlap_genes <- intersect(peak_genes, degs$gene_id)
# 功能富集(clusterProfiler)
library(clusterProfiler)
enrichGO(overlap_genes, OrgDb = org.Hs.eg.db,
ont = "BP", pAdjustMethod = "fdr")
3.2 单细胞表观组分析(scNanoATAC-seq2 案例)
清华大学纪家葵课题组开发的 scNanoATAC-seq2 技术,实现了单细胞水平的长读段染色质可及性分析,核心流程:
- 样本制备:单细胞核分离→透化处理→磁珠结合
- 文库构建:Tn5 转座酶切割→长片段富集→PCR 扩增
- 数据分析:
- 单细胞分群:Seurat 包基于峰矩阵聚类
- 谱系特异性调控元件鉴定:FindMarkers 识别差异可及性区域
- 父母本等位基因分析:利用 SNP 区分 X 染色体印记失活区域
四、常见问题与解决方案(避坑指南)
4.1 实验层面问题
| 问题现象 | 可能原因 | 解决方案 |
|---|---|---|
| ATAC-seq 线粒体 DNA 比例 > 30% | 细胞核分离不充分 | 优化裂解液配方,增加离心转速(1000g×10min) |
| CUT&Tag 峰信号弱 | 抗体亲和力低 | 选择 ChIP-grade 抗体,延长孵育时间至 16h |
| WGBS 转化效率 < 90% | 亚硫酸氢盐处理不充分 | 提高反应温度(55℃),延长处理时间(16h) |
4.2 分析层面问题
| 问题现象 | 解决方案 |
|---|---|
| 峰识别假阳性高 | 加入 Input 对照,调整 MACS2 q 值 cutoff(<0.01) |
| 差异分析样本重复性差 | 增加生物学重复(≥3 次),使用 DESeq2 的方差稳定化 |
| 长读段比对率低 | 采用 minimap2 比对,设置参数 -x map-ont |
五、工具资源汇总
5.1 核心工具链
| 分析步骤 | 推荐工具 | 优势 |
|---|---|---|
| 质控 | FastQC、ATACseqQC | 全面评估数据质量 |
| 比对 | BWA-MEM、minimap2 | 短读段 / 长读段适配 |
| 峰识别 | MACS2、HMMRATAC | 通用性 / 特异性兼顾 |
| 差异分析 | DiffBind、methylKit | 表观组专用统计模型 |
| 可视化 | IGV、ggplot2、UCSC Genome Browser | 交互式 / 批量绘图 |
5.2 数据库资源
- 参考基因组:UCSC Genome Browser、Ensembl
- 表观组数据:ENCODE、Roadmap Epigenomics
- 转录因子 Motif:JASPAR、HOMER Motif Database
总结与展望
表观基因组分析已从群体细胞水平迈向单细胞、长读段时代,scNanoATAC-seq2 等新技术的出现,为解析胚胎发育、疾病异质性等复杂生物学问题提供了强大工具。实操中需牢记 “实验设计为基础,数据质控为核心,多组学整合为方向” 的原则,灵活选择技术方案。未来,随着 AI 辅助的调控网络重构、空间表观组学技术的发展,表观基因组学将在精准医疗、再生医学等领域发挥更大作用。

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐
 4
4 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)