Datawhale组队学习-【AI4S】公开课 Lesson 1:流体场自编码器
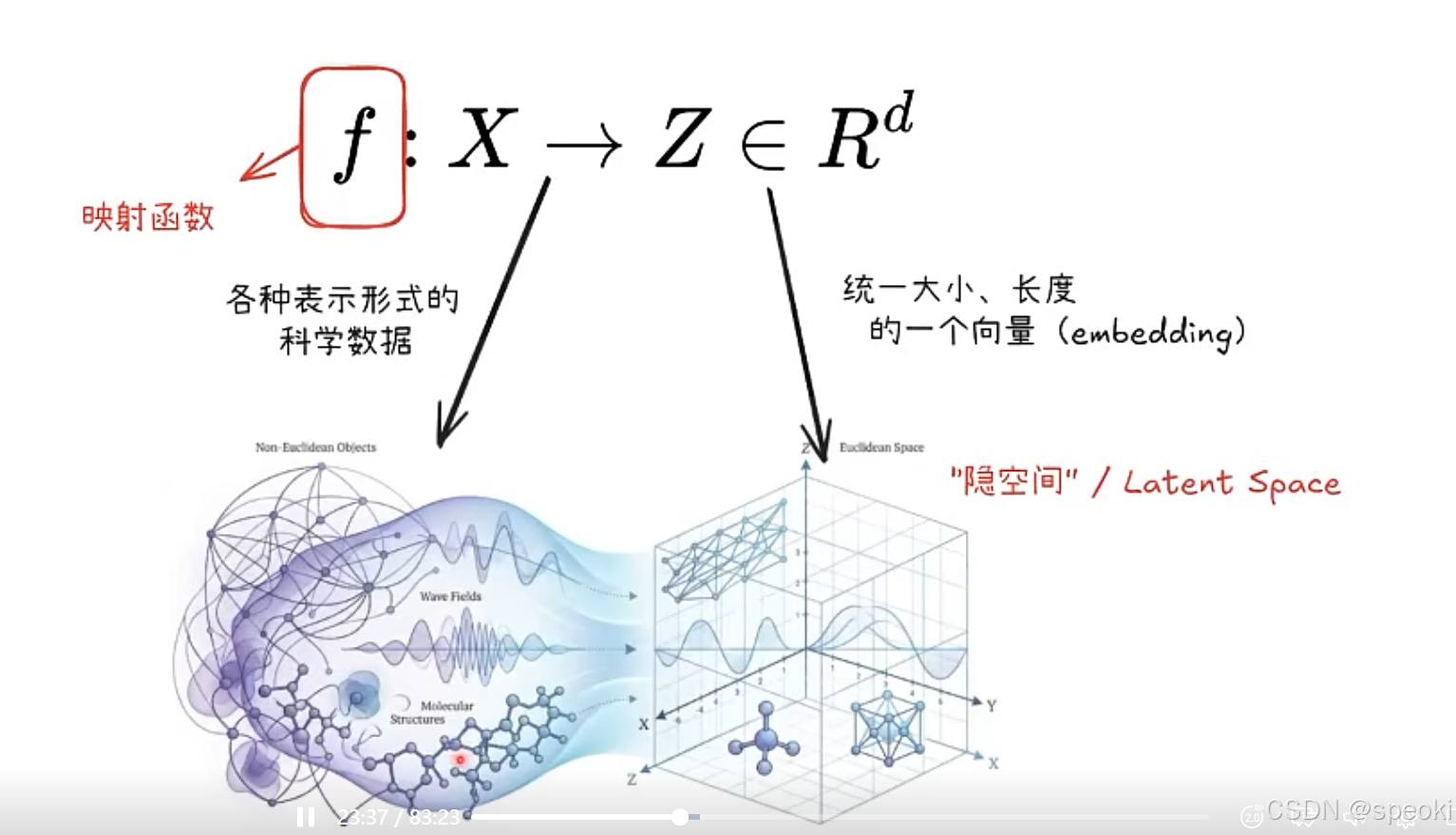
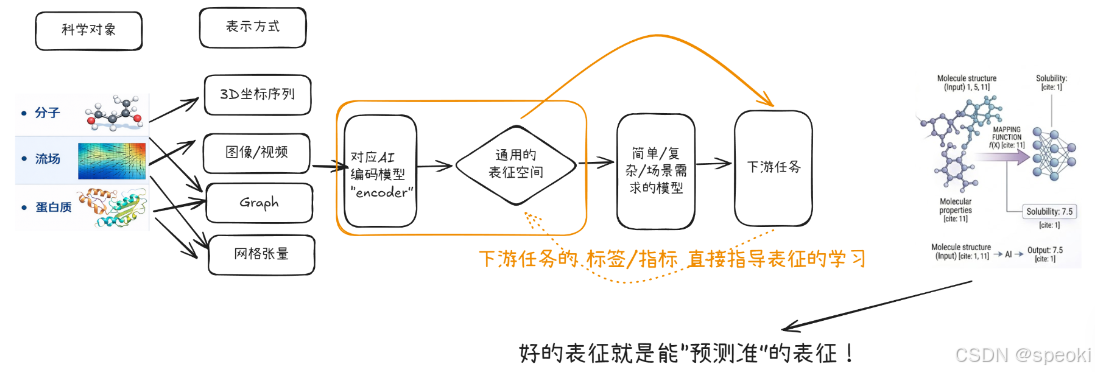
这张图里总结了通用的好的表征学习表征问题往往先于模型问题表征决定模型上限好的表征学习,就是把复杂的原始数据(图像、分子、文本等)映射到一个**既保持了原始结构(距离保持、连续性),又方便我们操作(可插值、可加性)**的向量空间,让后续的机器学习任务(分类、生成、预测)变得更容易、更鲁棒。两种典型的 “坏” 情况情况一:距离高估(Distance Overestimate)**现象:**两张几乎一样
https://github.com/xuangu-fang/AI4S-101/
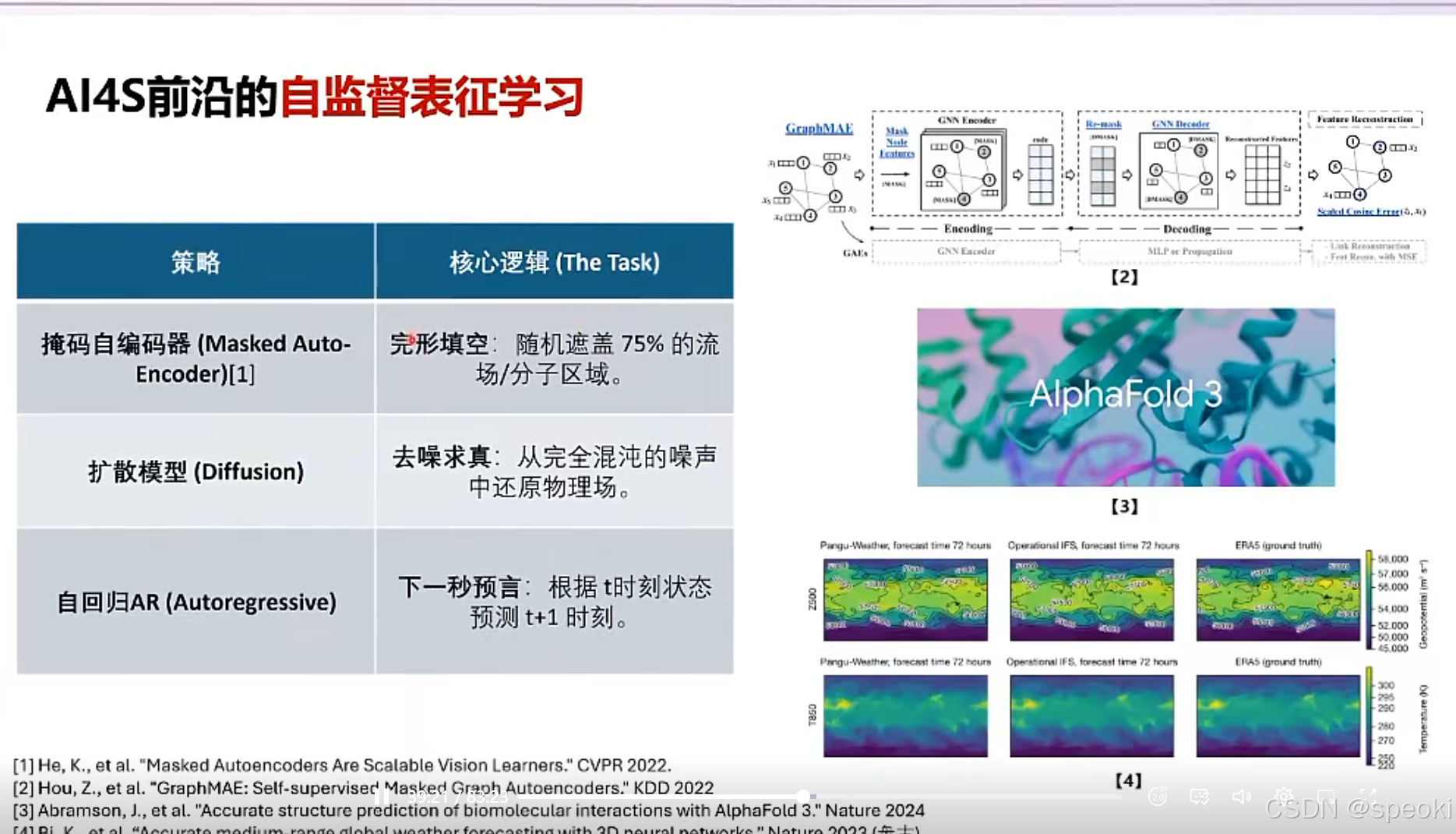
表征学习

AlphaFold 3
基于扩散模型架构,将蛋白质结构预测任务扩展到各类生物分子的结构和他们的相互作用上。
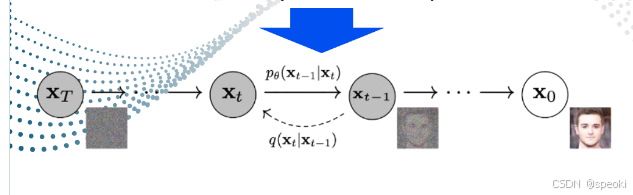
- 扩散模型怎么工作(类比理解)
你可以把它想象成:
先给一堆原子坐标加强噪声,变成一团混乱的 “原子雾”。
然后让模型一步步去噪,每次只修正一点点,直到所有原子都落到真实、稳定的三维位置。
最终直接输出每个原子的精确坐标,而不是先预测残基框架再推导原子。 - 相比 AlphaFold 2 的优势
通用性更强:不再依赖蛋白质特有的残基 / 侧链表示,能处理任意化学分子(DNA、RNA、小分子、离子等)。
生成更灵活:从 “按规则拼结构” 变成 “从噪声中生成结构”,更适合复杂、多分子的系统。
精度更高:在多分子复合物上,比传统物理方法和专用工具准确率提升约 50%。
AlphaFold 2 主要解决的是:“一条蛋白质序列 → 它的三维结构是什么”(单体蛋白)。而 AlphaFold 3 解决的是:“多种生物分子放在一起 → 它们各自长什么样、怎么结合、怎么相互作用”。
- 覆盖的分子类型(从 “单一蛋白” 到 “全生命分子”)
- 蛋白质(单体、多聚体、抗体 - 抗原)
- 核酸(DNA、RNA)
- 小分子配体(药物、辅酶、代谢物)
- 离子、金属、修饰残基等
- 核心能力:预测 “相互作用”
它不只是分别预测每个分子的结构,更能预测:
- 蛋白 - 蛋白怎么结合
- 蛋白 - DNA/RNA 怎么识别、结合
- 蛋白 - 小分子(药物)怎么对接、结合模式
- 多分子复合物(如核糖体、转录机器)的整体结构与作用方式
AlphaFold 3 用扩散模型,把 “预测单个蛋白结构” 升级成了 “统一预测所有生物分子及其复合物的结构与相互作用”,是从 “分子零件” 到 “分子机器” 的跨越。
扩展到相互作用是什么:
- 输入不是 “单一蛋白序列”,而是多分子的混合序列 / 结构信息
- 去噪过程中,模型会同时优化所有原子的相对位置—— 它不需要先区分 “哪个原子属于蛋白、哪个属于 DNA”,而是通过学习数据中的规律,让相互作用的原子(比如蛋白的结合位点氨基酸与 DNA 的碱基)自然 “靠近” 并形成稳定构象。
这个过程的本质是:在高维原子坐标空间中,寻找满足 “多分子结合稳定” 的能量最低点(对应拓扑空间里的 “最优流形”)。 - 多模态表征的统一嵌入:让不同分子 “能被模型理解”
不同生物分子的 “语言” 完全不同:蛋白是氨基酸序列,DNA/RNA 是碱基序列,小分子是化学式。要预测它们的相互作用,首先要让模型能把这些不同类型的信息转化为统一的特征。
- 多模态编码器
- 跨分子注意力机制:模型能计算注意力权重,重高的原子对,就是潜在的相互作用位点
扩散模型分子相互作用不是随机的,而是受物理化学法则支配,AlphaFold 3 把这些规则内置到模型训练和推理过程中
- 训练时,损失函数不仅包含 “预测坐标与真实坐标的误差”,还加入了物理约束项(比如原子间的距离不能小于范德华半径,避免原子重叠;氢键的键长要符合真实范围)。
- 推理时,模型生成的构象会自动满足这些约束 —— 比如带负电的 DNA 磷酸基团,会自然和带正电的蛋白氨基酸(如赖氨酸、精氨酸)结合,而不会和带负电的氨基酸靠近。
- 扩散反向采样:从随机噪声开始,每一步去噪都沿着势能面的梯度下降,自动趋向能量最低、约束满足的构象。
- Pairformer 预定义势能面:先计算所有原子对的pair 嵌入 z_ij,编码距离、电荷、氢键能力等约束,相当于在高维坐标空间画出 “允许区域”(边界条件)。
- 电荷 / 氢键自动匹配:模型在训练中已学习静电互补、氢键配对的统计规律,推理时会自然让:
- DNA 磷酸(负电)→ 蛋白 Lys/Arg(正电)靠近;
- 同电荷原子→ 排斥远离;
- 氢键供体(-OH/-NH)→ 受体(O/N)形成合理键长 / 角度。
- 这些约束相当于给高维坐标空间加了**“边界条件”**,让模型的去噪过程不会偏离真实的生物分子相互作用模式。
AlphaFold 2 需事后修复立体化学问题;AlphaFold 3 因约束内置到扩散生成,输出构象已基本无重叠、键长合理、相互作用符合物理。

扩散模型分成哪几个阶段
约束问题
什么是好的表征学习
这张图里总结了通用的好的表征学习的三个核心性质,我帮你逐一拆解,结合你熟悉的拓扑和向量空间视角来理解:
- 表征问题往往先于模型问题
- 表征决定模型上限
1. 距离保持(Distance Preservation)
- 直观理解:在原始空间里“相似”的东西,映射到表征向量空间后,它们的向量距离也应该“相近”;反之,原始空间里“不相似”的东西,表征向量也应该“离得远”。
- 数学表达:
d ( x i , x j ) ≈ ∥ f ( x i ) − f ( x j ) ∥ d(x_i, x_j) \approx \| f(x_i) - f(x_j) \| d(xi,xj)≈∥f(xi)−f(xj)∥
其中 d ( x i , x j ) d(x_i, x_j) d(xi,xj) 是原始数据空间的距离(比如图像的像素差异、分子的结构差异), f ( x ) f(x) f(x) 是表征映射函数, ∥ ⋅ ∥ \| \cdot \| ∥⋅∥ 是向量空间的范数(如欧氏距离)。 - 对应图中例子:椅子和凳子在视觉上相似,映射到嵌入空间后,它们的表征向量 z 1 z_1 z1 和 z 2 z_2 z2 也靠得很近,距离很小。
- 拓扑视角:这是在做一个近似等距映射,尽量保持原始数据空间的拓扑结构(邻域关系),避免“扭曲”数据的内在几何。
2. 连续性(Continuity)
- 直观理解:对原始数据做一个“小扰动”(比如给图片加一点噪声、改变分子的一个原子),得到的表征向量也只发生“小变化”,不会突然跳到完全不同的地方。
- 核心逻辑:小扰动 → 小变化。
- 对应图中例子:从猫(CAT)到狗(DOG)的插值路径上,每一步的变化都是平滑过渡的,表征向量的变化也是连续的,没有跳变。
- 拓扑视角:表征映射 f f f 是一个连续函数,保证了数据空间的邻域在表征空间中仍然是邻域,这对泛化和鲁棒性很重要。
3. 可插值/操作性/可加性
- 直观理解:表征向量空间是一个“有结构”的线性空间,你可以对向量做线性运算(插值、加减),得到的结果在语义上仍然有意义。
- 数学表达:
z = α z 1 + ( 1 − α ) z 2 z = \alpha z_1 + (1 - \alpha) z_2 z=αz1+(1−α)z2
其中 α ∈ [ 0 , 1 ] \alpha \in [0,1] α∈[0,1] 是插值系数, z z z 是介于 z 1 z_1 z1 和 z 2 z_2 z2 之间的新表征。 - 对应图中例子:
- 线性插值:从猫的表征 z cat z_{\text{cat}} zcat 到狗的表征 z dog z_{\text{dog}} zdog 做插值,中间的表征对应“像猫又像狗”的过渡形态。
- 向量加减:用“狗”的表征减去“四足动物”的表征,再加上“人”的表征,可能得到“人用两条腿走路”的语义操作。
- 拓扑视角:表征空间是一个线性流形,支持线性运算,这让我们可以像操作数字一样操作语义,实现生成、编辑等任务。
一句话总结
好的表征学习,就是把复杂的原始数据(图像、分子、文本等)映射到一个**既保持了原始结构(距离保持、连续性),又方便我们操作(可插值、可加性)**的向量空间,让后续的机器学习任务(分类、生成、预测)变得更容易、更鲁棒。
什么是坏的表征学习
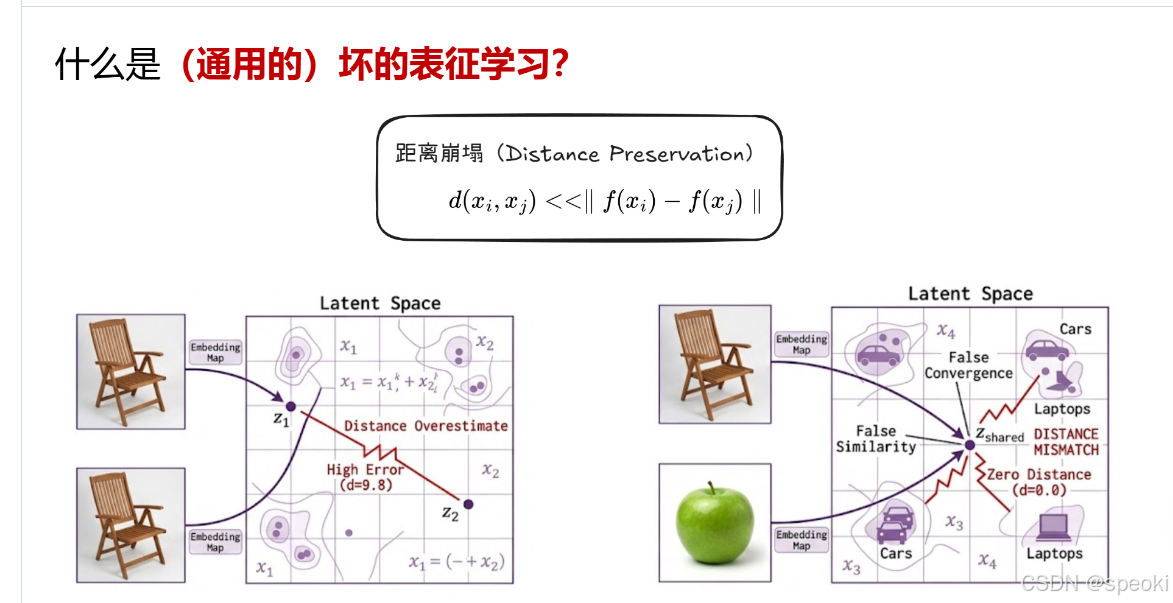

两种典型的 “坏” 情况
情况一:距离高估(Distance Overestimate)
**现象:**两张几乎一样的椅子(原始距离很小),被映射到表征空间后,它们的向量距离却非常大(图中 d=9.8)。
**后果:**模型会认为这两把椅子是完全不同的东西,导致分类、检索等任务完全失效。
**拓扑视角:**原始空间的邻域关系被完全撕裂,拓扑结构被严重扭曲。
情况二:距离错配(Distance Mismatch)
**现象:**一把椅子和一个苹果(原始空间里完全不相似),被映射到表征空间后,它们的向量距离几乎为零(d=0.0),甚至被错误地归到 “汽车” 和 “笔记本” 的簇里。
**后果:**模型产生 “虚假相似性”(False Similarity),把完全不相关的东西当成同类,导致严重的误判。
**拓扑视角:**不同的拓扑流形被错误地挤压到了同一个点,数据的内在结构完全丢失。
非连续性突变
理想情况:从猫(CAT)到狗(DOG)的表征插值路径是平滑、语义连贯的,中间每一步都对应 “像猫又像狗” 的合理过渡形态。
坏的情况:表征映射函数是不连续的。对原始数据做一个微小的扰动,表征向量却发生了剧烈跳变(Sudden Jumps)。
从猫到狗的插值路径中,出现了毫无意义的噪声、混乱的波形(Chaotic Wavefields)和物理扭曲(Physical Distortions)。
这导致了语义一致性的丧失(Loss of Semantic Consistency),中间的插值结果完全不可理解,与真实生物形态毫无关系。
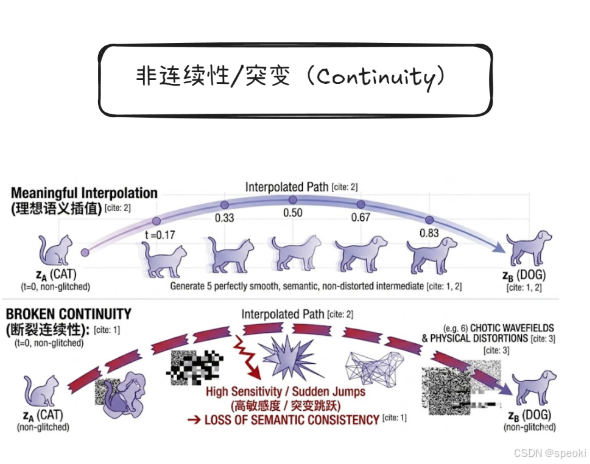
拓扑视角:表征映射 f f f 不再是连续函数,原始数据空间的邻域关系被破坏,导致模型对输入扰动极度敏感,鲁棒性极差。
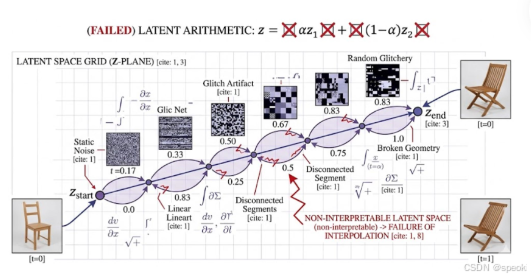
不可插值、语义缺失
- 理想情况:表征空间是线性的,向量运算(如 z = α z 1 + ( 1 − α ) z 2 ) z = \alpha z_1 + (1-\alpha)z_2) z=αz1+(1−α)z2))有明确的语义意义,比如“椅子”和“凳子”的插值可以得到“介于两者之间的家具”。
- 坏的情况:隐空间的插值和向量运算失去了实际意义:
- 从椅子( Z s t a r t Z_{start} Zstart)到另一把椅子( Z e n d Z_{end} Zend)的线性插值,中间出现了随机噪声(Random Glitchery)、伪影(Glitch Artifact)和不连续的片段(Disconnected Segments)。
- 整个隐空间的几何结构是破碎的(Broken Geometry),无法进行有意义的线性操作。
- 后果:无法通过向量运算进行语义编辑、生成或推理,表征空间变成了一个“黑箱”,失去了可操作性和可解释性。
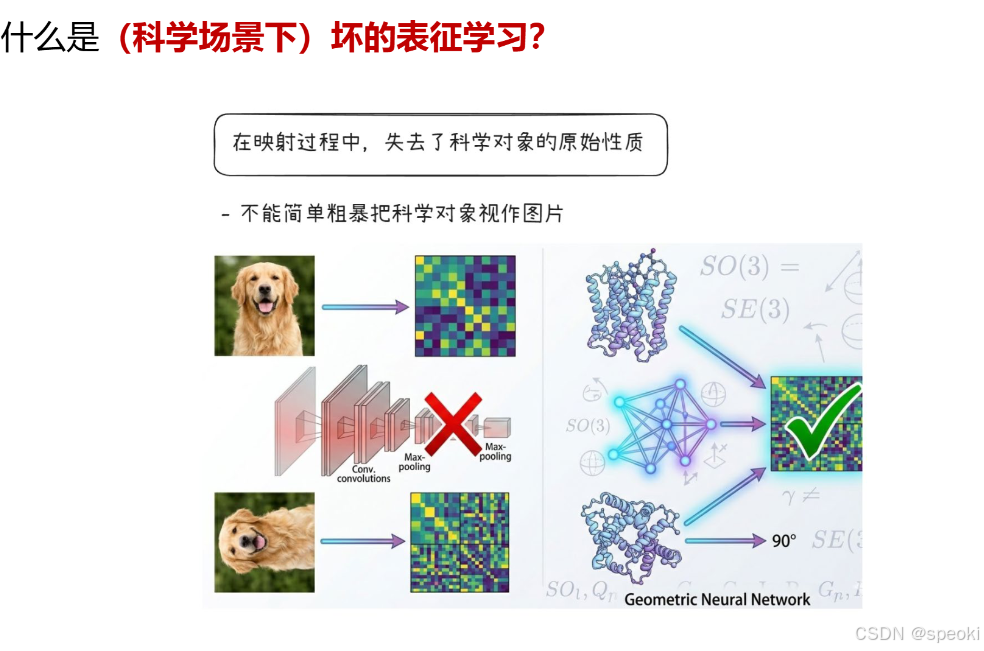
- 核心问题:丢失了科学对象的原始性质
在普通图像任务(比如识别狗)里,把图片旋转、翻转,用普通 CNN 提取特征,只要像素相似就能识别,不影响 “这是狗” 的语义。
但在科学场景下,对象的几何、拓扑、对称性、物理约束是其本质属性,一旦在表征映射中丢失,就会得到完全错误的结论。
比如蛋白质的三维结构、分子的手性、晶体的对称性,这些都不是 “视觉特征”,而是决定其功能的科学本质。
- 错误做法:把科学对象简单粗暴地当作图片处理
图中左侧展示了错误的做法:
把蛋白质结构直接当作 “3D 图片”,用普通 CNN 做卷积和池化。
这种做法完全无视了科学对象的几何与对称性,比如蛋白质的旋转、翻转会被当成不同的东西,或者被池化抹平关键结构。
结果就是表征丢失了科学意义,无法用于预测功能、相互作用等下游任务。
- 正确做法:用几何 / 等变神经网络(Geometric Neural Network)
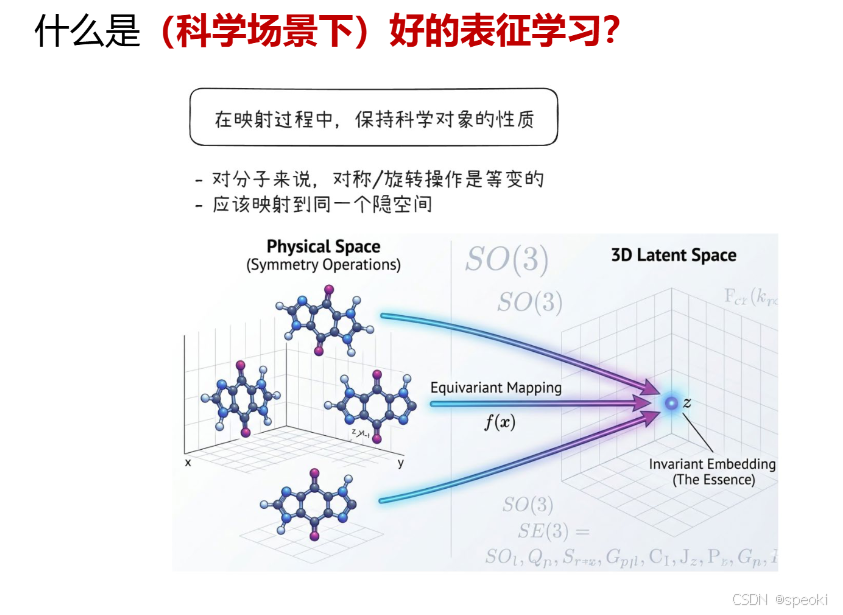
图中右侧展示了正确的思路:
使用几何神经网络(如等变 GNN),显式地建模科学对象的几何结构和对称性(如 SO(3)、SE(3) 群)。
当蛋白质旋转、平移时,表征也会按照对应的群操作进行变换,而不是被破坏。
这样得到的表征保留了科学对象的几何与拓扑性质,才能真正用于科学发现(如预测蛋白功能、分子相互作用)。
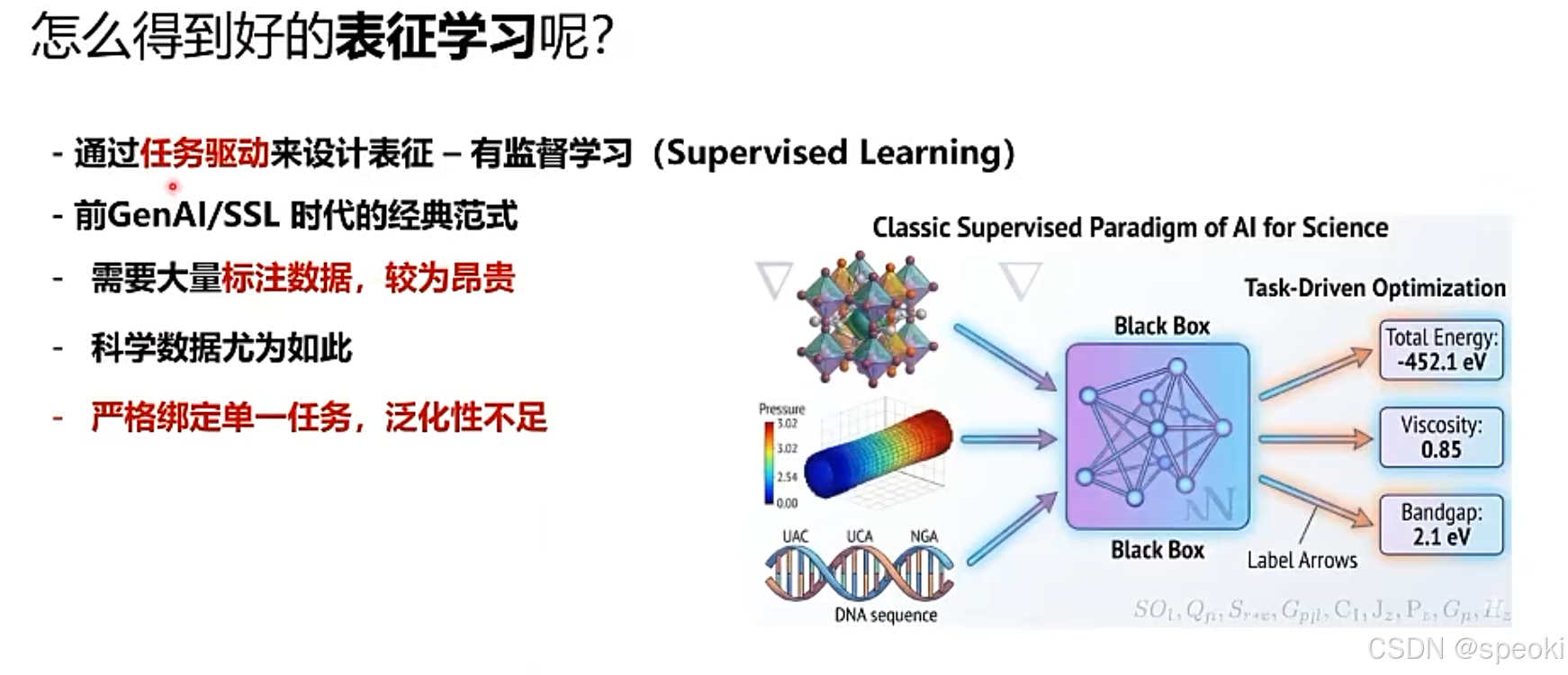
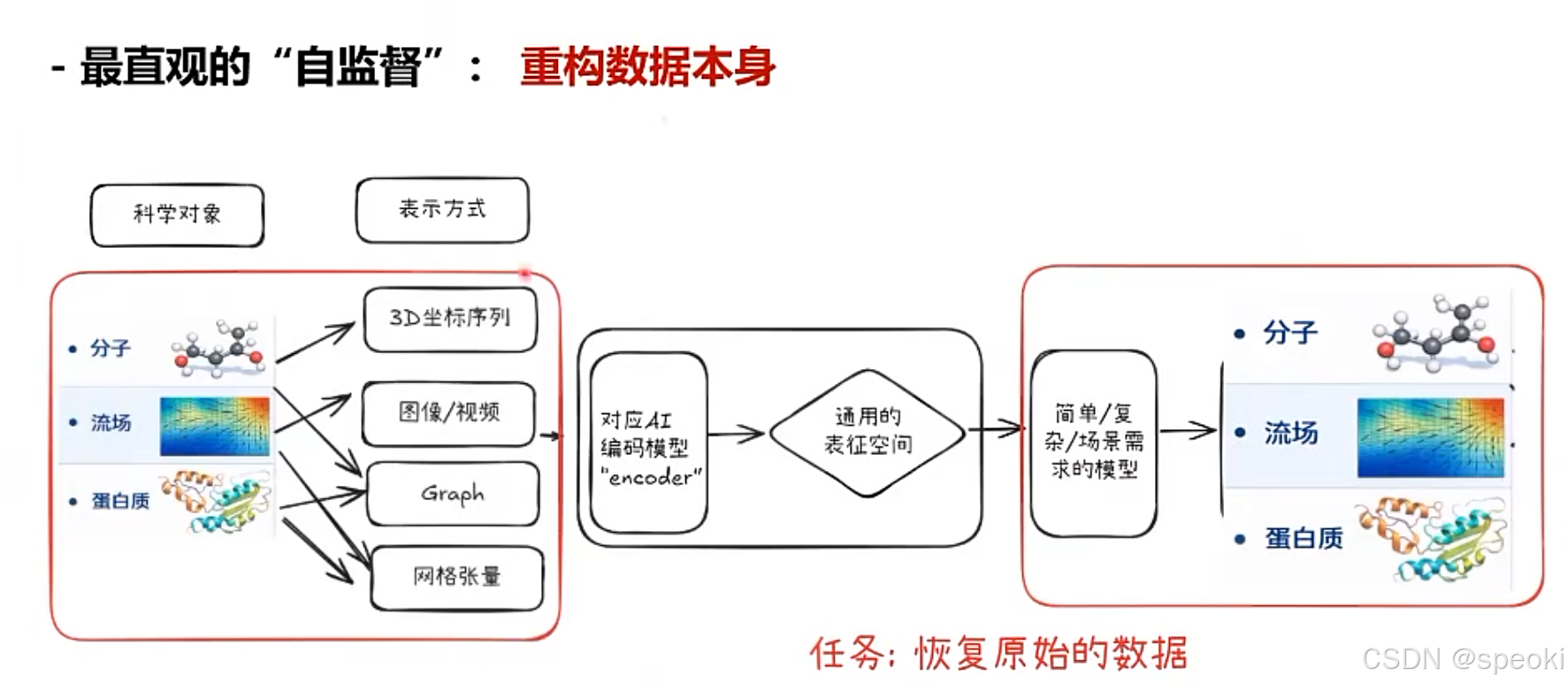
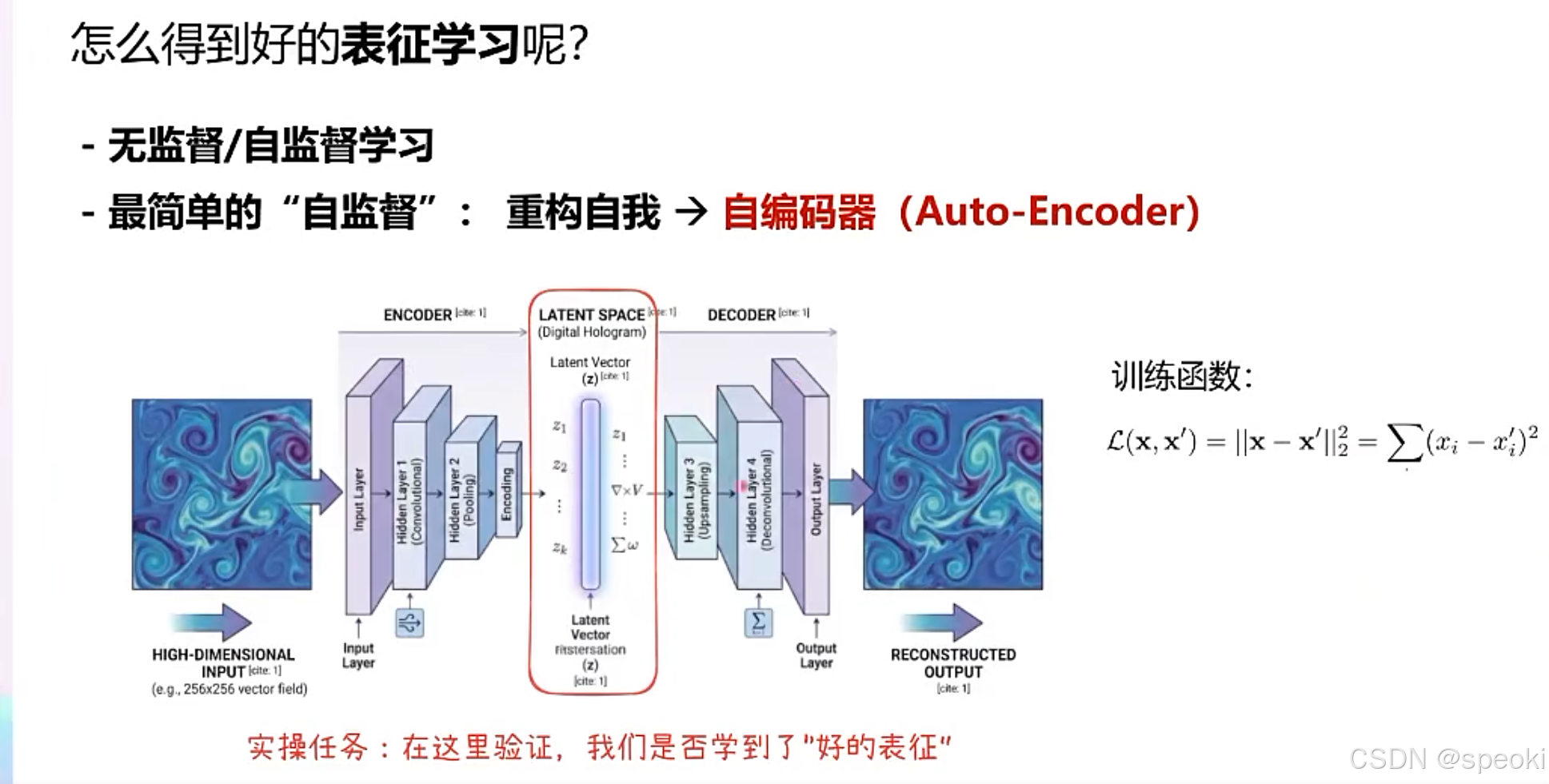
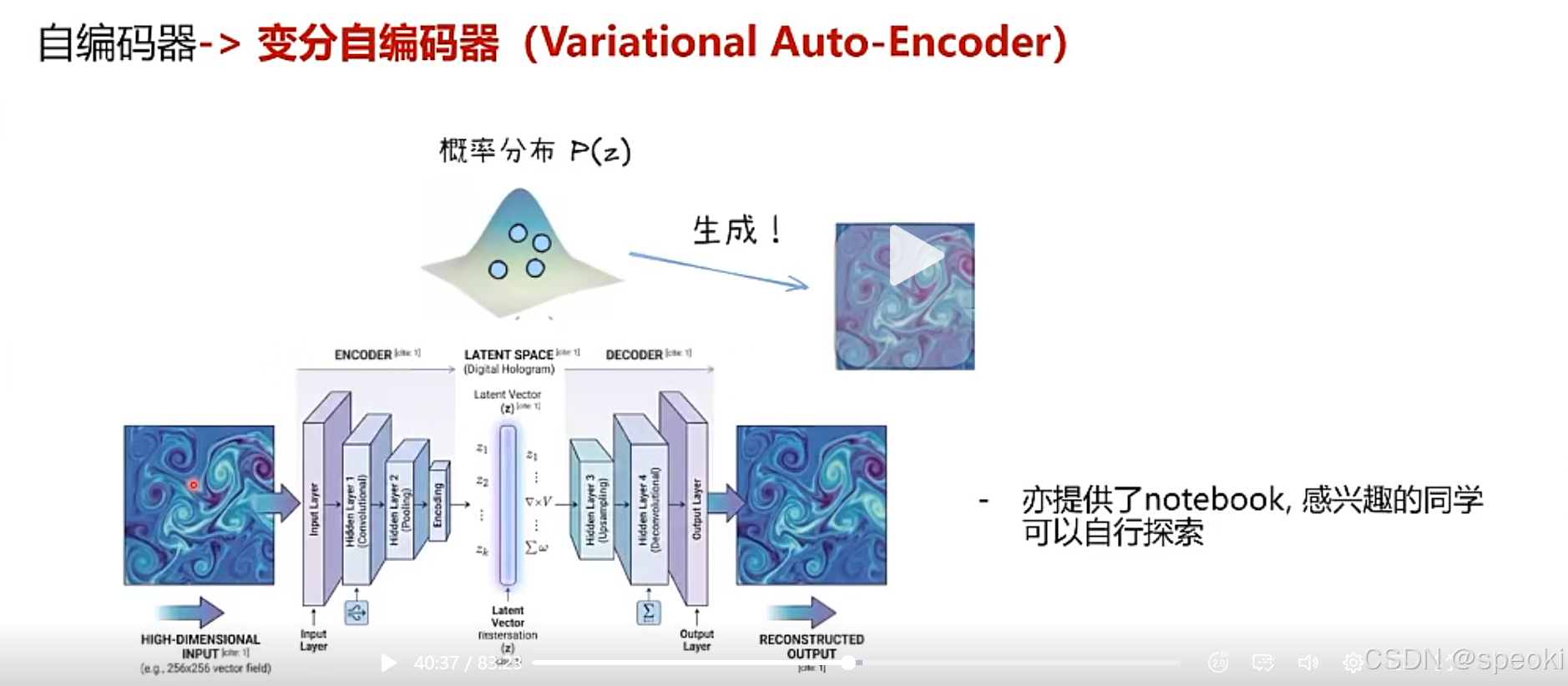
怎么得到好的表征学习呢?
自回归AR



有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐


 0
0 0
0- 0



 已为社区贡献19条内容
已为社区贡献19条内容

所有评论(0)