fastGEO V1.9.0: R语言+AI,快速获取想要的GEO数据集
想要学习和使用有时间看看以前写的教程:R包开发-fastGEO,快速下载、注释GEO芯片表达谱和临床数据fastGEO v1.01,快速下载GEO表达谱、差异分析、火山图、热图fastGEO v1.03,让手动构建GEO注释文件更快捷fastGEO V1.6.1 这个版本强的可怕,GEO数据自动下载、探针注释、Shiny AppfastGEO v1.7.0 大更新,支持PCA、差异分析、火山图、热
-
最新版本: 1.9.0
-
更新日期: 20260208
-
可以对着视频教程学习和使用:R包fastGEO-GEO数据库快速下载探针注释
-
没有安装包的需要付费获取安装包:
- 直接添加微信转账100, 发送fastR和fastGEO的压缩包
- 以前是20, 老用户依然可免费获取
- 很多资料后面都会慢慢涨价, 早买会便宜很多!
- 购买后可进入交流群,后续版本更新和问题答疑都在群里进行

R包介绍
- 想要学习和使用有时间看看以前写的教程:
- R包开发-fastGEO,快速下载、注释GEO芯片表达谱和临床数据
- fastGEO v1.01,快速下载GEO表达谱、差异分析、火山图、热图
- fastGEO v1.03,让手动构建GEO注释文件更快捷
- fastGEO V1.6.1 这个版本强的可怕,GEO数据自动下载、探针注释、Shiny App
- fastGEO v1.7.0 大更新,支持PCA、差异分析、火山图、热图、差异箱线图、去批次等分析
- fastGEO V1.8.0 快速下游分析,多数据集批量下载处理,火山图优化
为什么开发这个包
-
你在下载和分析GEO数据库或者使用GEOquery包时是否遇到以下问题?
- getGEO总是下载文件失败
- 网页中series文件或者补充文件下载总是失败
- 无法确定表达矩阵是否已经log化
- 不会进行探针ID到基因SYBBOL的注释转换
- GPL注释文件下载总是中断
- GPL注释文件不包含基因SYMBOL
- GPL注释文件的SYMBOL存在一对多和多对一的情况
- GPL注释文件仅能查看但没有下载按钮
- 下载的表达谱基因包含日期
- 检索到很多GSE数据集, 但是逐个看太费时间
-
如果有以上这些问题, 那么我开发的R包fastGEO大概率能解决你的问题!
实现的功能
- 一行代码实现对芯片表达谱数据的下载、标准化、样本间矫正、探针注释、去除重复基因、恢复日期基因等等操作
- 最终你会直接得到行名是基因列名是样本的标准化之后的表达谱, 以及对应的样本临床信息
- 探针注释是fastGRO开发的主要目的, 可以实现对多种情况的GPL平台进行下载和处理
- 对于文件下载失败的问题提供多种解决办法
- 提供简单的下游分析和可视化快捷函数, 如PCA、差异分析、火山图、热图、差异箱线图、去批次等分析
- 支持多个数据集批量进行下载与注释
- 快速执行PCA、差异分析、火山图、热图, 展示合并图, 快速了解数据情况
- V1.9更新自动检索数据集并提取数据集信息, 结合AI极大地提高数据集筛选效率
适用场景
- 首先, GEO网站在国外, 下载网速受限制是难以避免的, 会科学上网会更好一点, 但偶尔还是下载失败, 需要多尝试
- fastGEO下载表达谱是针对于芯片数据, 不支持高通量数据, 因为NGS数据不存放在series里, 而是在补充文件里
- 探针注释能够成功的前提是GPL平台要提供了注释文件, 并且文件包含基因ID信息
- SYMBOL/ENSEMBL/ENTREZ/REFSEQ等等都可以
- 如果没有提供基因信息, 那也不能无中生有
- 对于任何测序类型, 都支持一行代码获取样本临床信息
功能演示
- 安装在下一节介绍
rm(plot_boxplot)
Warning message in rm(plot_boxplot):
"object 'plot_boxplot' not found"
# 加载R包
library(fastGEO)
Loading required package: fastR
Loading required package: ggplot2
Warning message:
"package 'ggplot2' was built under R version 4.4.3"
Loading required package: GEOquery
Loading required package: Biobase
Loading required package: BiocGenerics
Attaching package: ‘BiocGenerics’
The following objects are masked from ‘package:stats’:
IQR, mad, sd, var, xtabs
The following objects are masked from ‘package:base’:
anyDuplicated, aperm, append, as.data.frame, basename, cbind, colnames, dirname, do.call, duplicated, eval, evalq, Filter, Find,
get, grep, grepl, intersect, is.unsorted, lapply, Map, mapply, match, mget, order, paste, pmax, pmax.int, pmin, pmin.int,
Position, rank, rbind, Reduce, rownames, sapply, setdiff, table, tapply, union, unique, unsplit, which.max, which.min
Welcome to Bioconductor
Vignettes contain introductory material; view with 'browseVignettes()'. To cite Bioconductor, see 'citation("Biobase")', and for
packages 'citation("pkgname")'.
Setting options(‘download.file.method.GEOquery’=‘auto’)
Setting options('GEOquery.inmemory.gpl'=FALSE)
Loading required package: limma
Attaching package: ‘limma’
The following object is masked from ‘package:BiocGenerics’:
plotMA
The following object is masked from ‘package:fastR’:
strsplit2
Loading required package: AnnoProbe
AnnoProbe v 0.1.0 welcome to use AnnoProbe!
If you use AnnoProbe in published research, please acknowledgements:
We thank Dr.Jianming Zeng(University of Macau), and all the members of his bioinformatics team, biotrainee, for generously sharing their experience and codes.
Loading required package: stringr
Loading required package: httr
Attaching package: ‘httr’
The following object is masked from ‘package:Biobase’:
content
Loading required package: XML
Loading required package: openxlsx
Warning message:
"package 'openxlsx' was built under R version 4.4.2"
Loading required package: dplyr
Attaching package: ‘dplyr’
The following object is masked from ‘package:Biobase’:
combine
The following objects are masked from ‘package:BiocGenerics’:
combine, intersect, setdiff, union
The following object is masked from ‘package:fastR’:
case_when
The following objects are masked from ‘package:stats’:
filter, lag
The following objects are masked from ‘package:base’:
intersect, setdiff, setequal, union
Loading required package: rvest
========================================
欢迎使用 fastGEO (版本 1.9.0)
- 作者: 生信摆渡
- 微信: bioinfobaidu1
- 帮助文档: help(package = fastGEO)
========================================
完整流程
# 为了演示, 先将处理好的文件删除
final_file = "test/00_GEO_data_GSE18105/GSE18105_GPL570_annoted.RData"
if(file.exists(final_file)) unlink(final_file)
# invisible(sapply(list.files("../../functions", full.names = T), source))
# 下载数据
obj = download_GEO("GSE18105", out_dir = "test/00_GEO_data_GSE18105")
>>>>> 识别到本地注释文件, 将优先使用!
>>>>> getGEO2已经执行过, 将跳过, 设置'force_getGEO2 = TRUE'强制重新运行!
>>>>> 执行探针-基因注释, 平台: GPL570 ...
>>>>> 使用R包内置的注释文件...
>>>>> 去除重复的基因注释, 使用方法: max...
>>>>> 处理结束!
# 获取分组
# 注意这里尽量先指定实验组再指定对照组, 因为后续分析在版本1.8.0之后会默认将第一个分组作为实验组进行分析
group_list = get_GEO_group(obj, group_name = "characteristics_ch1.1", "cancer, homogenized" = "Cancer", "normal, homogenized" = "Normal")
group = group_list$group
expM = group_list$expM
SID = group_list$SID
table(group)
group
Cancer Normal
17 17
# PCA
set_image(6, 6) # 仅在jupyter里使用, 其他软件可忽略
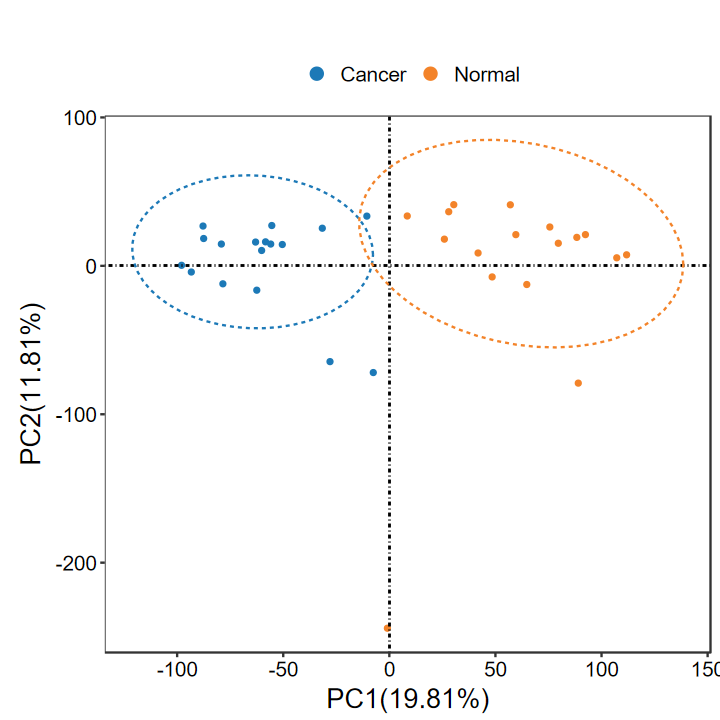
run_PCA(expM, group)
Warning message:
"[1m[22mUsing `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
[36mℹ[39m Please use `linewidth` instead."
# 加载本地函数, 用于我做测试, 用户不用运行
# invisible(sapply(list.files("../../functions", full.names = T), source))
# 差异分析
DEG_tb = run_DEG_limma(expM, group = group, Case = "Cancer", Control = "Normal", out_name = "test/00_GEO_data_GSE18105/GSE18105_DEG")
Warning message:
"Parameter 'Case' is deprecated since fastGEO 1.8.0, please use 'case' instead!"
Warning message:
"Parameter 'Control' is deprecated since fastGEO 1.8.0, please use 'control' instead!"
Number of Up regulated genes: 1006/2102(47.86%)
Number of Down regulated genes: 1096/2102(52.14%)
Number of Not Change genes: 20779/22881
- 版本1.8.0之后参数名均改为小写了, 但是仍保留首字母大写的写法, 不会报错只会提醒你, 记得改一下吧
DEG_tb = run_DEG_limma(expM, group = group, case = "Cancer", control = "Normal", out_name = "test/00_GEO_data_GSE18105/GSE18105_DEG")
Number of Up regulated genes: 1006/2102(47.86%)
Number of Down regulated genes: 1096/2102(52.14%)
Number of Not Change genes: 20779/22881
- 而且这个版本也不需要指定分组, 会默认将第一类分组作为实验组, 这在get_GEO_group的时候有提到过, 自己要清楚默认情况对不对
DEG_tb = run_DEG_limma(expM, group, out_name = "test/00_GEO_data_GSE18105/GSE18105_DEG")
Not specify 'case' and 'control', will detect automatically!
!!! Please make sure contrast: Cancer vs. Normal
Number of Up regulated genes: 1006/2102(47.86%)
Number of Down regulated genes: 1096/2102(52.14%)
Number of Not Change genes: 20779/22881
- 也增加了差异基因的比例和结果报告的输出, 更方便写文章了
readLines("test/00_GEO_data_GSE18105/GSE18105_DEG_report.txt")
‘结果表明,与Normal对照组相比,Cancer组共有2,102个差异表达基因,其中上调有1,006(47.86%),下调有1,096(52.14%)’
# 加载本地函数, 用于我做测试, 用户不用运行
# invisible(sapply(list.files("../../functions", full.names = T), source))
# 火山图
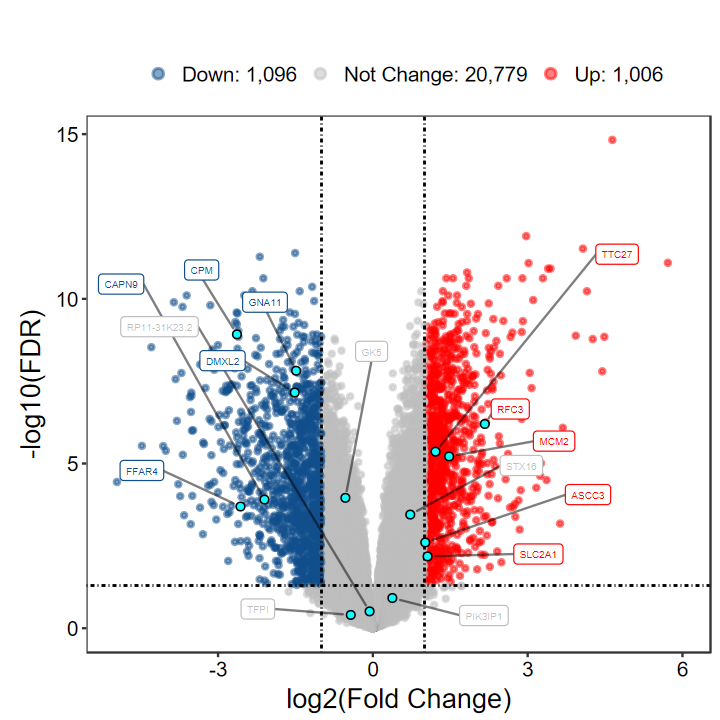
plot_volcano_limma(DEG_tb, out_name = "test/00_GEO_data_GSE18105/GSE18105_volcano")
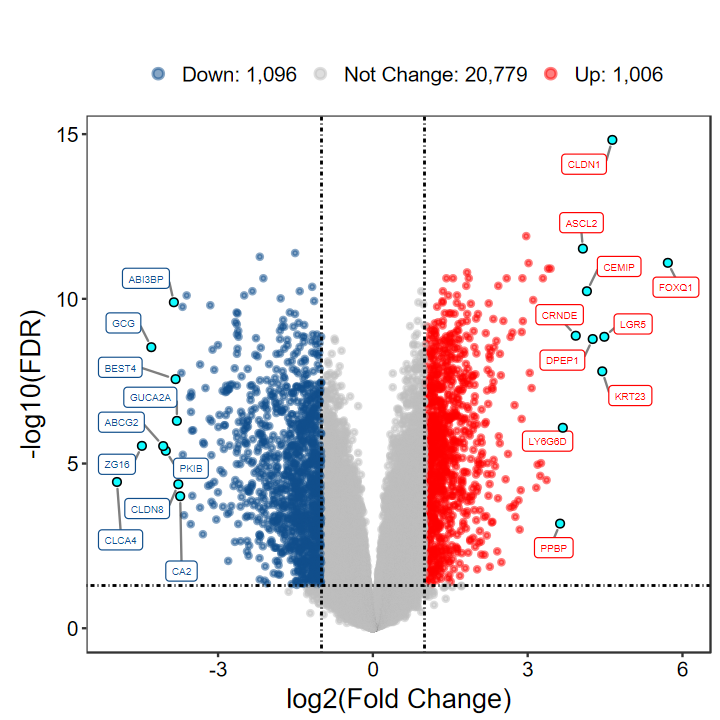
- 版本1.8.0的火山图也更新了一点, 对指定标注的基因的点进行强调显示
- 点的color和fill可以通过label.point.color和label.point.fill进行自定义
# 火山图
plot_volcano_limma(DEG_tb, out_name = "test/00_GEO_data_GSE18105/GSE18105_volcano", label.point.color = "green", label.point.fill = "yellow")
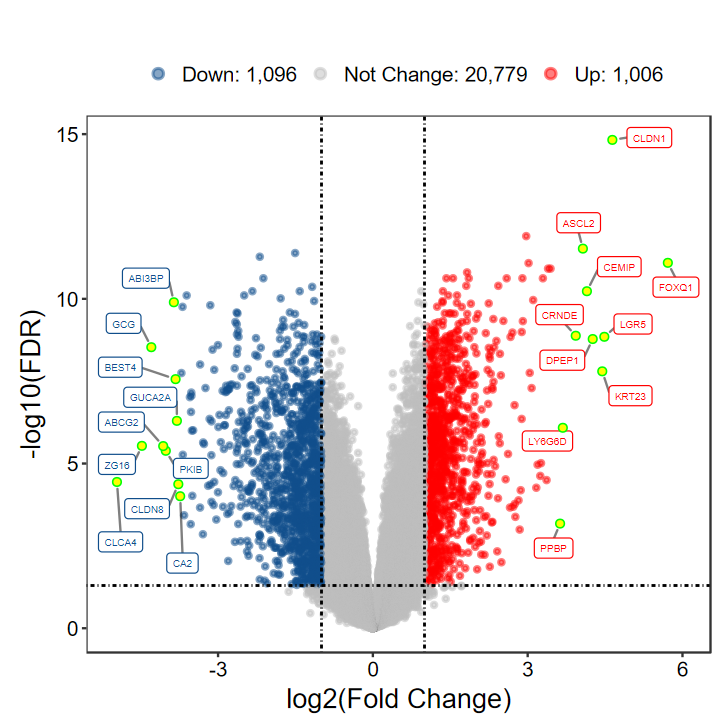
# 觉得标签的太挤?可通过xlims和ylims调整
plot_volcano_limma(DEG_tb, out_name = "test/00_GEO_data_GSE18105/GSE18105_volcano", xlims = c(-6, 6))
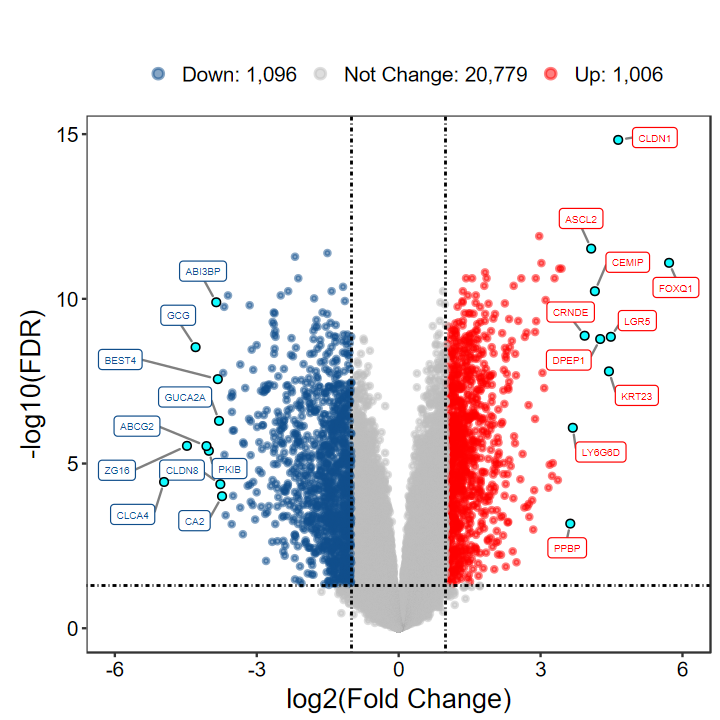
# 想自己指定基因?指定label即可
# 自定义label基因, 每组随机挑选5个
set.seed(1234)
gene_select = as.character(aggregate(rownames(DEG_tb), by = list(DEG_tb$DEG), function(x) sample(x, 5))[, 2])
gene_select
- 'CPM'
- 'STX16'
- 'TTC27'
- 'FFAR4'
- 'RP11-31K23.2'
- 'MCM2'
- 'GNA11'
- 'PIK3IP1'
- 'RFC3'
- 'CAPN9'
- 'TFPI'
- 'ASCC3'
- 'DMXL2'
- 'GK5'
- 'SLC2A1'
plot_volcano_limma(DEG_tb, out_name = "test/00_GEO_data_GSE18105/GSE18105_volcano", label = gene_select)
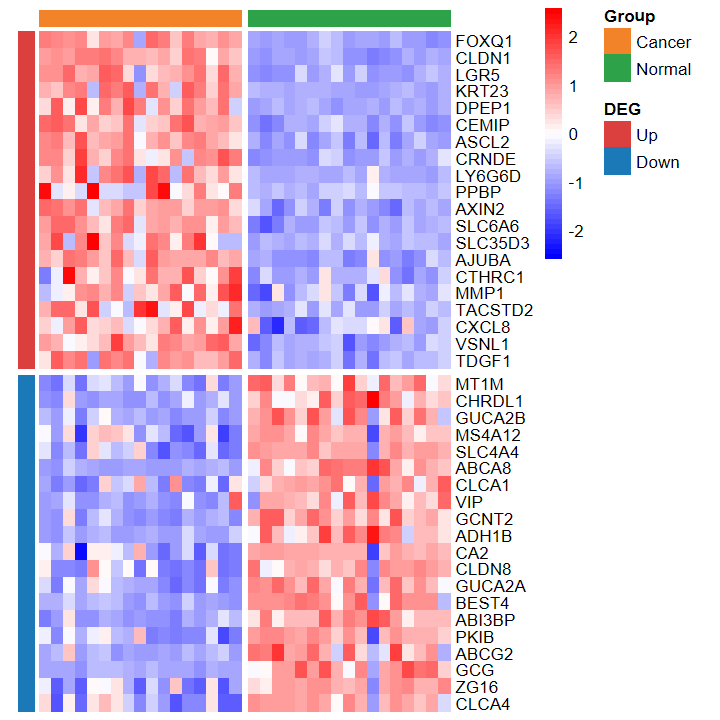
# 热图
plot_heatmap_DEG(expM, DEG_tb, group)
# detach("package:fastGEO", unload = TRUE)
# 查看帮助文档, 在RStudio里可以点击查看每一个函数的帮助文档
help(package = fastGEO)
Documentation for package 'fastGEO'
Information on package ‘fastGEO’
Description:
Package: fastGEO
Title: Quickly download and annotate GEO data chip datasets
Version: 1.9.0
Authors@R: person("Jiahao", "Wang", , "wjhyyds@sina.cn", role = c("aut", "cre"), comment = c(ORCID = "YOUR-ORCID-ID"))
Description: You can use the fastqGEO to easily download GEO chip expression profiles and clinical data and automate probe-gene
annotation.
License: `use_mit_license()`, `use_gpl3_license()` or friends to pick a license
Encoding: UTF-8
Roxygen: list(markdown = TRUE)
RoxygenNote: 7.3.2
Depends: R (>= 3.5.0), fastR (>= 1.5.0), ggplot2 (>= 3.5.2), GEOquery (>= 2.72.0), limma (>= 3.60.4), AnnoProbe (>= 0.1.0),
stringr (>= 1.5.1), httr (>= 1.4.7), XML (>= 3.99.0.17), openxlsx (>= 4.2.7.1), dplyr (>= 1.1.4), rvest (>= 1.0.4)
Imports: data.table, ggplot2, GEOquery, limma, readr
Suggests: org.Hs.eg.db, org.Mm.eg.db, clusterProfiler
LazyData: true
NeedsCompilation: no
Packaged: 2026-02-08 09:25:42 UTC; Jia
Author: Jiahao Wang [aut, cre] (YOUR-ORCID-ID)
Maintainer: Jiahao Wang <wjhyyds@sina.cn>
RemoteType: local
RemoteUrl: W:\02_Study\R_build\build\02_fastGEO\version\1.9.0\fastGEO_1.9.0.tar.gz
Built: R 4.4.1; ; 2026-02-08 09:25:44 UTC; windows
Index:
analyse_GEO Comprehensive GEO Expression Data Analysis Pipeline
anno_GEO Annotate GEO Expression Matrix
anno_GEO_AnnoProbe Annotate GEO Data Using AnnoProbe Package
anno_GEO_online Annotate GEO Data Using Online Annotation
anno_NGS Annotate NGS Expression Matrix
anno_obj GEO Platform Annotation Reference Object
check_expr_log_normalized Automatically detect and normalize log transformation status of expression profile
count_to_exp Convert Counts to Normalized Expression
data_date_to_gene Convert table for date to gene convertion
date_to_gene Convert Date-Based IDs to Gene Symbols
download_GEO Download and Process GEO Dataset
download_GEO_file Download GEO Files (Series Matrix or Supplementary Files)
download_GEO_multi Download and Process GEO Dataset multiply
download_GEO_series_multi Download GEO series multiply
extract_GPL_anno Extract Gene Symbol Annotations from GEO Platform (GPL) Data
extract_GSE_info_single Extract Metadata from Single GSE HTML File
getGEO2 Download and Process GEO Dataset
get_GEO_group Create Sample Groups from GEO Data
get_GEO_pheno Get GEO Phenotype Data
kill_curl Force Terminate All curl Processes
plot_boxplot Create Boxplots for Gene Expression
plot_heatmap_DEG Heatmap of DE Genes
plot_volcano_limma Volcano Plot for DE Results
read_GPL Read GEO Platform (GPL) File
read_GPL_file Read and Process GPL Annotation File
read_GPL_url Download and Process GPL Annotation from GEO Website
read_gds_result Read GSE Accessions from GDS Result TXT
run_ComBat Batch Effect Correction with ComBat
run_DEG_deseq2 Differential Expression Analysis with DESeq2
run_DEG_limma Differential Expression Analysis with limma
run_PCA Perform and Visualize Principal Component Analysis
run_fastGEO_app 启动 fastGEO 的 Shiny 应用
search_GEO Search GEO, Retrieve GSE Accessions, and Extract Metadata
table_GEO_clinical Tabulate Clinical Characteristics from GEO Sample Annotations
- 每一个函数的帮助文档可以使用
?函数名进行自行查看, 比如?download_GEO
analyse_GEO
- 快速执行PCA、差异分析、火山图、热图, 展示合并图, 快速了解数据情况
# 加载本地函数, 用于我做测试, 用户不用运行
# invisible(sapply(list.files("../../functions", full.names = T), source))
set_image(15, 6)
analyse_GEO(expM, group, out_name = "test/00_GEO_data_GSE18105/GSE18105_pipeline")
Not specify 'case' and 'control', will detect automatically!
!!! Please make sure contrast: Cancer vs. Normal
Step1. PCA ...
Step2. DEG ...
Number of Up regulated genes: 1006/2102(47.86%)
Number of Down regulated genes: 1096/2102(52.14%)
Number of Not Change genes: 20779/22881
Step3. volcano ...
Step4. heatmap ...
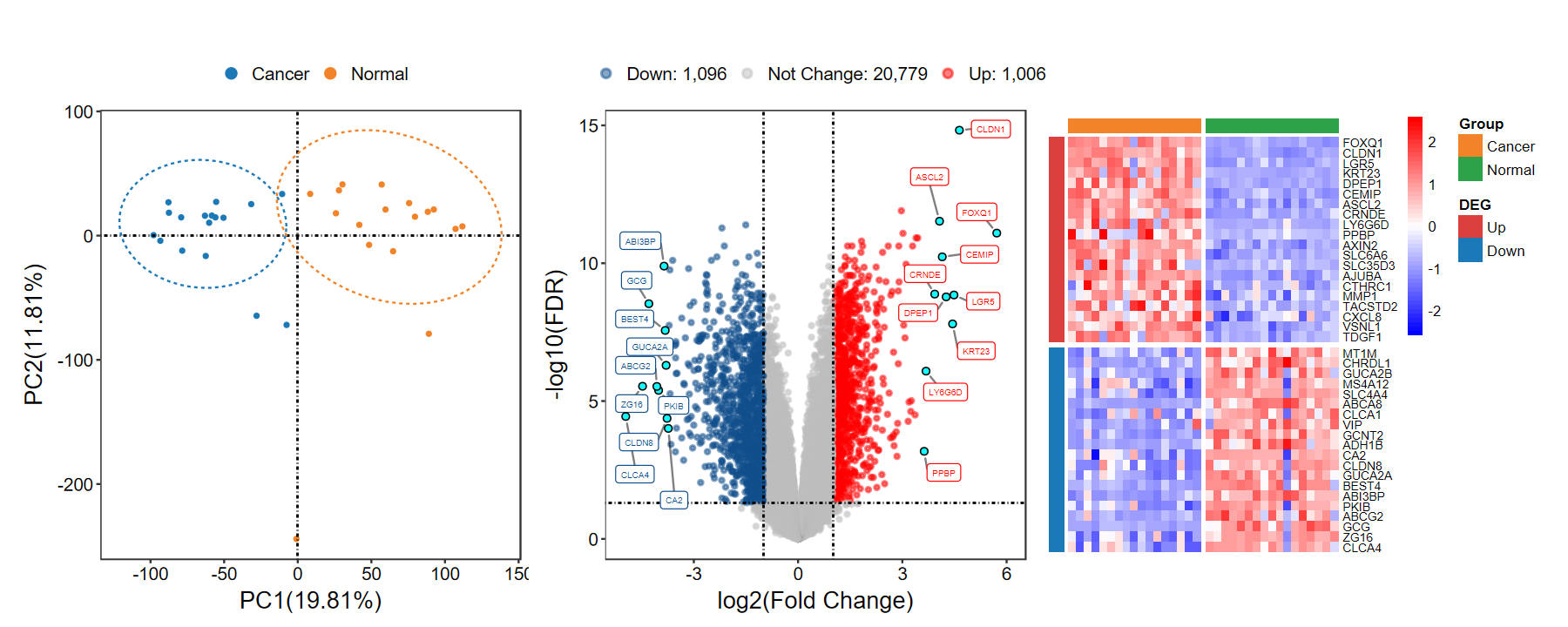
download_GEO
- 主函数, 输入GSE号, 实现自动化下载和处理数据
# 查看参数
args(download_GEO)
function (GSE, out_dir = "00_GEO_data", anno_file = NULL, method = "max",
timeout = 3600 * 24, download_method = "getGEO", force = FALSE,
force_getGEO2 = FALSE, symbol_name = NULL)
NULL- GSE: GSE号
- out_dir: 文件保存目录, 默认为当前目录的00_GEO_data文件夹, 可自行修改
- anno_file: 注释文件, 现在已经内置了注释文件, 可以不用指定
- method: 重复基因的处理方法
- max: 默认, 取最大表达量做为该基因的表达水平, 相对较快且合理
- mean: 取均值, 太慢
- remove: 直接去除重复基因, 最快, 但不推荐
- sum: 求和, 用的比较少, 暂不推荐
- download_method: 下载文件的方式, 如果存在本地curl则用本地的, 不存在则使用getGEO的curl
- V1.9更新: download_method默认为getGEO, 即不使用curl进行下载, 因为GEO数据库不允许这样了
- symbol_name: 注释文件中含有基因信息的那一列的列名
# 这种下载文件的方式可能也比较慢, 看运气
# 如果下载失败, 看下面的download_GEO_file函数
obj = download_GEO("GSE294904", out_dir = "test/00_GEO_data_GSE294904")
>>>>> 访问数据集: GSE294904...
>>>>> 下载尝试次数: 1 ... FALSE
下载成功!
>>>>> 处理测序平台: GPL570
>>>>> 识别到6个样本, 34个注释信息
>>>>> 输出样本注释表至: test/00_GEO_data_GSE294904/GSE294904_GPL570_sample_anno.csv
Attaching package: ‘xlsx’
The following objects are masked from ‘package:openxlsx’:
createWorkbook, loadWorkbook, read.xlsx, saveWorkbook, write.xlsx
>>>>> 识别到数据未log化, 执行log(expM+1)
>>>>> 执行样本间矫正...
>>>>> 处理成功文件保存至: test/00_GEO_data_GSE294904/GSE294904_GPL570_raw.RData
>>>>> 执行探针-基因注释, 平台: GPL570 ...
>>>>> 使用R包内置的注释文件...
>>>>> 去除重复的基因注释, 使用方法: max...
>>>>> 处理结束!
# 结果包含以下文件
cbind(dir("test/00_GEO_data_GSE294904/"))
| GSE294904.RData |
| GSE294904_GPL570.RData |
| GSE294904_GPL570_annoted.RData |
| GSE294904_GPL570_expM.csv |
| GSE294904_GPL570_expM_annoted.csv |
| GSE294904_GPL570_Normalization.pdf |
| GSE294904_GPL570_raw.RData |
| GSE294904_GPL570_sample_anno.csv |
| GSE294904_GPL570_sample_anno.xlsx |
| GSE294904_series_matrix.txt.gz |
GSE294904_GPL570.RData是最终保存的文件, 也是返回的obj的内容, 可以load进行使用sample_anno是样本临床信息, 可打开快速浏览和筛选样本
# 保存的对象
names(obj)
- 'GPL'
- 'expM'
- 'expM_raw'
- 'sample_anno'
obj$GPL
‘GPL570’
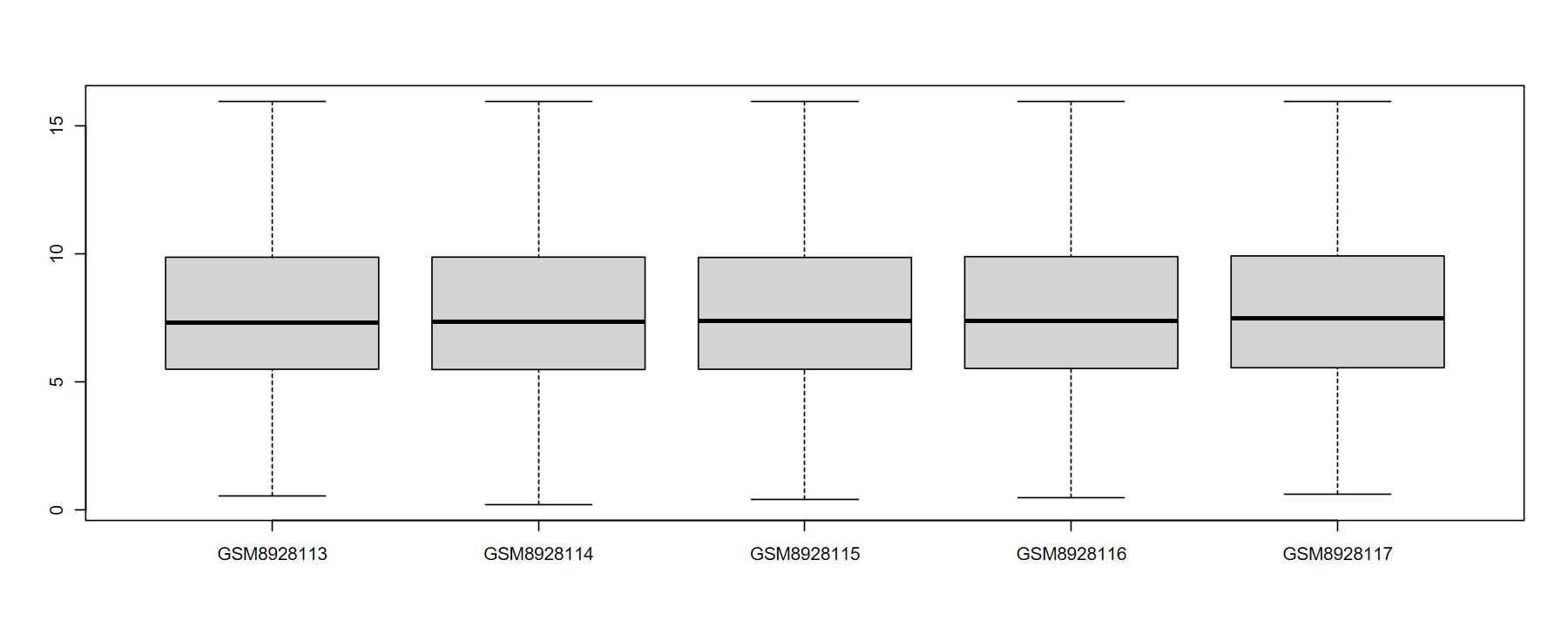
# 完美的表达谱
hd(obj$expM)
Type: data.frame Dim: 22881 × 6
GSM8928113 GSM8928114 GSM8928115 GSM8928116 GSM8928117
A1BG 7.664415 7.834532 8.045193 8.715155 8.702467
A1BG-AS1 4.182379 6.486369 3.594159 4.888389 5.456329
A1CF 3.311157 6.515058 6.397253 7.288225 6.671245
A2M 5.580024 6.934858 5.895105 6.041618 6.338498
A2M-AS1 2.398807 5.313860 5.330637 5.697162 5.843399
boxplot(obj$expM[, 1:5])
# 样本信息
hd(obj$sample_anno)
Type: data.frame Dim: 6 × 34
title geo_accession status submission_date last_update_date
GSM8928113 NCI-ADR-RES control GSM8928113 Public on Apr 30 2025 Apr 17 2025 Apr 30 2025
GSM8928114 NCI-ADR-RES treated GSM8928114 Public on Apr 30 2025 Apr 17 2025 Apr 30 2025
GSM8928115 GCS control GSM8928115 Public on Apr 30 2025 Apr 17 2025 Apr 30 2025
GSM8928116 GCS treated GSM8928116 Public on Apr 30 2025 Apr 17 2025 Apr 30 2025
GSM8928117 asGCS control GSM8928117 Public on Apr 30 2025 Apr 17 2025 Apr 30 2025
- 如果提供的数据集是高通量数据, 则只会返回样本注释, 表达谱需要自己去补充文件里下载处理
sample_anno = download_GEO("GSE173955")
>>>>> 访问数据集: GSE173955...
>>>>> 下载尝试次数: 1 ... FALSE
下载成功!
>>>>> 处理测序平台: GPL18460
>>>>> 识别到18个样本, 43个注释信息
>>>>> 输出样本注释表至: 00_GEO_data/GSE173955_GPL18460_sample_anno.csv
>>>>> 未识别到表达谱, 提供的数据集可能是高通量测序, 请确认!
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE173955
>>>>> 高通量数据的表达谱请自行从补充文件下载处理!
>>>>> 将仅返回样本注释表!
hd(sample_anno)
Type: data.frame Dim: 18 × 43
title geo_accession status submission_date last_update_date
GSM5283449 Alzheimer's Disease_Biological replicate 1 GSM5283449 Public on Dec 23 2021 May 05 2021 Dec 23 2021
GSM5283450 Alzheimer's Disease_Biological replicate 2 GSM5283450 Public on Dec 23 2021 May 05 2021 Dec 23 2021
GSM5283451 Alzheimer's Disease_Biological replicate 3 GSM5283451 Public on Dec 23 2021 May 05 2021 Dec 23 2021
GSM5283452 Alzheimer's Disease_Biological replicate 4 GSM5283452 Public on Dec 23 2021 May 05 2021 Dec 23 2021
GSM5283453 Alzheimer's Disease_Biological replicate 5 GSM5283453 Public on Dec 23 2021 May 05 2021 Dec 23 2021
download_GEO_file
- 专门用来下载文件的
- 如果download_GEO下载series文件失败, 可先尝试这种方式, 然后再运行download_GEO
- 还可以用来下载所有的补充文件或其中一个
# 如果download_GEO下载文件失败, 可下面这样专门下载series文件
download_GEO_file("GSE294904", out_dir = "test/00_GEO_data_GSE294904/")
All file already downloaded in test/00_GEO_data_GSE294904/
# 显示已经下载好了, 因为上面的download_GEO我下载成功了, 嘿嘿
# 我们可以换个输出目录再演示一下
download_GEO_file("GSE294904", out_dir = "test/00_GEO_data_GSE294904-2/")
All file already downloaded in test/00_GEO_data_GSE294904-2/
# 结果包含以下文件
cbind(dir("test/00_GEO_data_GSE294904-2/"))
| GSE294904_series_matrix.txt.gz |
# 有时候还是比较慢的, 很离谱, 不过直接在浏览器下载却出奇的快
# 所以也支持跳转到浏览器进行下载, 逆天!
# 设置 method = "browser" 就行了
# 同时也可以设置输出目录, 程序会自动粘贴输出目录, 然后在浏览器里直接复制这个目录, 进行保存文件
# 所以浏览器的文件下载要设置每次询问下载位置
download_GEO_file("GSE294904", out_dir = "test/00_GEO_data_GSE294904-4/", method = "browser")
All file already downloaded in test/00_GEO_data_GSE294904-4/
# 很快啊, 几秒就完成了
cbind(dir("test/00_GEO_data_GSE294904-3/"))
| GSE294904_RAW.tar |
| GSE294904_series_matrix.txt.gz |
# 也可以用来下载补充文件
download_GEO_file(GSE = "GSE294904",
file = "GSE294904_RAW.tar",
out_dir = "test/00_GEO_data_GSE294904-3/",
method = "browser")
All file already downloaded in test/00_GEO_data_GSE294904-3/
download_GEO_series_multi
- 批量从浏览器下载series文件, 因为浏览器下载比在R里下载要稳定很多很多, 所以又提供了这个快捷函数
# 在浏览器下载series文件
datasets = c("GSE179285", "GSE75214", "GSE87466")
download_GEO_series_multi(datasets, out_dir = "test/download_GEO_series_multi")
All file already downloaded in test/download_GEO_series_multi
All file already downloaded in test/download_GEO_series_multi
All file already downloaded in test/download_GEO_series_multi
download_GEO_multi
- 同样的, 可以提供多个GSE批量下载、预处理、注释, 其中下载建议采用上面的浏览器的方法
- 另一个能正常运行的重要前提就是注释这一步能够通过, 如果不能处理注释的话, 还是需要使用后续的一系列的方法先解决然后再次批量运行
download_GEO_multi(datasets, out_dir = "test/download_GEO_series_multi")
>>>>> 准备并行下载...
-
结果很快就出来了, 这里不是循环处理, 而是并行处理, 同时对每个数据集执行download_GEO操作, 速度快很多!
-
后面的V1.8.0就没有更新的内容了, 跟老版本一样
-
更新维护这个R包真的消耗我巨大的精力, 但是看到能让数据分析方便很多很多, 成就感还是满满的!
get_GEO_pheno
- 快速获取样本临床信息
- V1.9之后, 可以舍弃这个函数了, 主函数download_GEO将兼容这个功能!
# 芯片数据
sample_anno = get_GEO_pheno("GSE294904", out_dir = "test/00_GEO_data_GSE294904")
>>>>> 访问数据集: GSE294904...
>>>>> 下载尝试次数: 1 ... FALSE
下载成功!
>>>>> 处理测序平台: GPL570
>>>>> 识别到6个样本, 34个注释信息
>>>>> 输出样本注释表至: test/00_GEO_data_GSE294904/GSE294904_GPL570_sample_anno.csv
>>>>> 识别到数据未log化, 执行log(expM+1)
>>>>> 执行样本间矫正...
>>>>> 处理成功文件保存至: test/00_GEO_data_GSE294904/GSE294904_GPL570_raw.RData
hd(sample_anno)
Type: NULL Dim: 0
NULL
# 其他测序类型也支持
sample_anno = get_GEO_pheno("GSE121950", out_dir = "test/00_GEO_data_GSE121950")
>>>>> 访问数据集: GSE121950...
>>>>> 下载尝试次数: 1 ... FALSE
下载成功!
>>>>> 处理测序平台: GPL11154
>>>>> 识别到6个样本, 41个注释信息
>>>>> 输出样本注释表至: test/00_GEO_data_GSE121950/GSE121950_GPL11154_sample_anno.csv
>>>>> 未识别到表达谱, 提供的数据集可能是高通量测序, 请确认!
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE121950
>>>>> 高通量数据的表达谱请自行从补充文件下载处理!
>>>>> 将仅返回样本注释表!
hd(sample_anno)
Type: data.frame Dim: 6 × 41
title geo_accession status submission_date last_update_date
GSM3450401 villus_induced abortion_rep1 GSM3450401 Public on Nov 27 2018 Oct 29 2018 Nov 27 2018
GSM3450402 villus_induced abortion_rep2 GSM3450402 Public on Nov 27 2018 Oct 29 2018 Nov 27 2018
GSM3450403 villus_induced abortion_rep3 GSM3450403 Public on Nov 27 2018 Oct 29 2018 Nov 27 2018
GSM3450404 villus_recurrent spontaneous abortion_rep1 GSM3450404 Public on Nov 27 2018 Oct 29 2018 Nov 27 2018
GSM3450405 villus_recurrent spontaneous abortion_rep2 GSM3450405 Public on Nov 27 2018 Oct 29 2018 Nov 27 2018
getGEO2
-
用于下载和预处理数据的函数
-
不过这个函数不需要直接使用, 因为download_GEO和get_GEO_pheno会进行自动调用
-
这里主要讲解下过程和原理
-
getGEO2有默认的下载方式, 其中curl是更推荐的一种方式, 但仍存在超时的情况, V1.9之后已舍弃curl下载的功能
-
所以我这里优先使用本地的curl进行下载, 如果你还没有安装则需安装一下, 并添加系统环境变量
-
下载完后也会自动保存样本临床信息
-
自动判断是否需要进行log2(expM + 1)处理
-
自动进行样本间矫正
-
最终返回临床信息和矫正后的表达谱, 但此时探针ID还没有进行注释
# 相当于 download_GEO 的中间步骤
obj = getGEO2("GSE294904", out_dir = "test/00_GEO_data_GSE294904")
>>>>> 访问数据集: GSE294904...
>>>>> 下载尝试次数: 1 ... FALSE
下载成功!
>>>>> 处理测序平台: GPL570
>>>>> 识别到6个样本, 34个注释信息
>>>>> 输出样本注释表至: test/00_GEO_data_GSE294904/GSE294904_GPL570_sample_anno.csv
>>>>> 识别到数据未log化, 执行log(expM+1)
>>>>> 执行样本间矫正...
>>>>> 处理成功文件保存至: test/00_GEO_data_GSE294904/GSE294904_GPL570_raw.RData
obj$GPL
NULL
hd(obj$expM)
Type: NULL Dim: 0
NULL
hd(obj$sample_anno)
Type: NULL Dim: 0
NULL
read_GPL 系列
- 下载完之后就需要进行探针ID注释了, 这里是最麻烦的一步, 因为探针注释文件格式千变万化 -_-
- 所以第一步就是下载和读取GPL注释文件
- 这几个函数也是提供给大家, 可以自行构建注释表, 因为总注释表我更新肯定不及时, 而且平台太多了, 总是遇到没有注释过的平台
read_GPL
- 直接读取本地的GPL文件, 这种情况前提是你要能下载下来
- 先不做处理
# https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GPL570
# 下载至 test/GPL570/GPL570-55999.txt
GPL570_raw = read_GPL("test/GPL570/GPL570-55999.txt")
hd(GPL570_raw)
Type: data.frame Dim: 54675 × 16
ID GB_ACC SPOT_ID Species Scientific Name Annotation Date
1 1007_s_at U48705 Homo sapiens Oct 6, 2014
2 1053_at M87338 Homo sapiens Oct 6, 2014
3 117_at X51757 Homo sapiens Oct 6, 2014
4 121_at X69699 Homo sapiens Oct 6, 2014
5 1255_g_at L36861 Homo sapiens Oct 6, 2014
extract_GPL_anno
- 从读取好的注释数据框里提取探针ID列和包含基因名的列
- 这是最麻烦的一点, 这一列有很多不同的写法, 我们总结了以下几类列名
colnames_SYMBOL = c("Gene symbol", "Gene Symbol", "GENE_SYMBOL", "GeneSymbol", "Symbol", "SYMBOL", "gene_assignment")
colnames_ENTREZ = c("ENTREZ_GENE_ID")
colnames_REFSEQ = c("GB_ACC")
colnames_ENSEMBL = c("GENE_ID", "SPOT_ID")
colnames_candicate = c(colnames_SYMBOL, colnames_ENTREZ, colnames_REFSEQ, colnames_ENSEMBL)
- 只要存在这些列就说明是可以注释的, 只需要使用
bitr函数进行ID转换就行了
load2("./test/00_GEO_data_GSE294904/GSE294904.RData")
Loading objects:
GSE294904
GPL = GSE294904$GPL
GPL
‘GPL570’
expM_raw = GSE294904$expM_raw
hd(expM_raw)
Type: matrix Dim: 5 × 5
GSM8928113 GSM8928114 GSM8928115 GSM8928116 GSM8928117
1007_s_at 10.014355 10.135966 11.008001 11.071630 10.268858
1053_at 11.633831 11.610780 11.346647 11.354608 10.919310
117_at 6.661878 6.857272 4.313413 3.496718 5.036547
121_at 10.278252 10.507507 10.323595 10.588387 9.398323
1255_g_at 2.666673 3.210481 3.041658 5.902110 2.303889
GPL570_anno = extract_GPL_anno(GPL570_raw)
Found annotation column: Gene Symbol
hd(GPL570_anno)
Type: data.frame Dim: 54675 × 2
ID SYMBOL
1 1007_s_at DDR1
2 1053_at RFC2
3 117_at HSPA6
4 121_at PAX8
5 1255_g_at GUCA1A
- 这样我们就得到了ID转换表了, 已经成功一大半了
read_GPL_file
- 从本地GPL读取注释文件(read_GPL实现)
- 随后使用extract_GPL_anno提取探针ID列和包含基因名的列
- 返回包含两列的注释表: ID列和SYMBOL列
# 读取和提取一步到位
anno_tb = read_GPL_file("./test/GPL570/GPL570-55999.txt", out_dir = "test/GPL570")
Processing: GPL570 ...
Found annotation column: Gene Symbol
Annotation file Save to C:/software/R-4.4.1/library/fastGEO/data/anno_obj.RData
hd(anno_tb[[1]])
Type: data.frame Dim: 54675 × 2
ID SYMBOL
1 1007_s_at DDR1
2 1053_at RFC2
3 117_at HSPA6
4 121_at PAX8
5 1255_g_at GUCA1A
- 成功生成注释表后, 将自动与总注释表进行合并
- 这也是这里返回的是列表的原因, 因为总的注释数据就是一个列表, 每个元素就是一个平台的注释数据框
- 合并完之后会自动保存到fastGEO的安装目录下的data目录
- 下次就不需要再为这个平台准备注释文件了, 因为download_GEO 默认首先会从这里读取总注释数据
- 所以自己遇到我还没整理的平台可以运行一下这几个函数, 成功后再次运行 download_GEO可能就好了
read_GPL_url
- 很多小伙伴会遇到一个问题, 就是GPL注释文件虽然可以查看, 但是不能下载
- 而且查看久了浏览器会卡死, 因为几万行的数据浏览器是扛不住的
- 后来我发现这个查看的网页的注释内容是固定且完整的, 所以将网页下载下来并提取注释表格就行了
- 嘿, 我真他娘的天才!还真可以!
- 你只需要快速将网页的链接粘贴下来, 然后就能关闭网页了, 不然等会浏览器就崩溃了
- 拿到链接之后直接扔给
read_GPL_url就能获取处理完之后的注释表格了 - 所以这里包含从HTML提取注释表格并执行
extract_GPL_anno两个步骤
url = "https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?view=data&acc=GPL10379&id=5393&db=GeoDb_blob45"
anno_tb = read_GPL_url(url, out_dir = "test/GPL1037")
Processing: GPL10379 ...
File downloaded successfully!
Found annotation column: ORF
New annotation file save to: C:/software/R-4.4.1/library/fastGEO/data/anno_obj.RData
hd(anno_tb[[1]], 10:20, 2)
Type: data.frame Dim: 52378 × 2
ID SYMBOL
20 100147785_TGI_at
21 100138290_TGI_at CEP350
22 100135090_TGI_at
23 100310799_TGI_at
24 100123722_TGI_at
25 100311433_TGI_at
26 100137544_TGI_at
27 100142142_TGI_at
28 100129675_TGI_at
29 100307002_TGI_at
30 100144445_TGI_at SEMA6A
anno_GEO 系列
- 那问题又来了, 如果平台没有提供GPL注释文件或不包含基因名信息怎么办?
- 好在我发现还有其他的获取注释文件的方法
- anno_GEO_AnnoProbe: Jimmy Zeng写的R包, 整理了很多R包的注释信息, 尤其是将一些只提供碱基序列的探针给比对了一遍
- anno_GEO_online: 少数情况下, 有些GSE自带注释文件, 可以直接在线下载读取
- 所以我们有三种注释的方式, 自己准备注释文件(或者我的包已经自带的)、AnnoProbe和online三种
anno_GEO_AnnoProbe
- 使用AnnoProbe包获取注释信息
anno_GEO_online
- 使用GSE里的在线注释文件获取注释信息
anno_GEO
- 准备好注释文件后就可以对表达谱进行注释了
- 由于存在多个探针对应同一个基因的情况, 所以需要进行去重
- 前面有讲过, 通过method参数指定重复基因的处理方法
- max: 默认, 取最大表达量做为该基因的表达水平, 相对较快且合理
- mean: 取均值, 太慢
- remove: 直接去除重复基因, 最快, 但不推荐
- sum: 求和, 用的比较少, 暂不推荐
anno_NGS
- 这个是独立的功能, 用于对行名是ENSEMBL ID的二代测序表达谱的注释
- 二代测序表达谱数据一般存放在补充文件
date_to_gene
- 最后还有一个功能, 就是我发现有些表达谱的基因名是日期, 不排除是Excel搞的鬼: R语言-将日期恢复为基因
- 所以最后再检查以下是否包含日期名, 如果有则自动进行转换
run_fastGEO_app
- 最后还有个更逆天的功能, 你可以在浏览器执行这些操作
- 其实就是使用Shiny基于我的这些函数搭建的一个分析平台
- 其实我并不会Shiny的语法, 主要是是ChatGPT帮我写的, 我稍微改了一下
- 大家可以体验一下😅, 目前还是要在R里面操作, 更灵活一点, 这个是搞着玩的暂时就是
# 执行下面的函数打开Shiny App
# 我这里就不运行了, 只展示截图
# run_fastGEO_app()
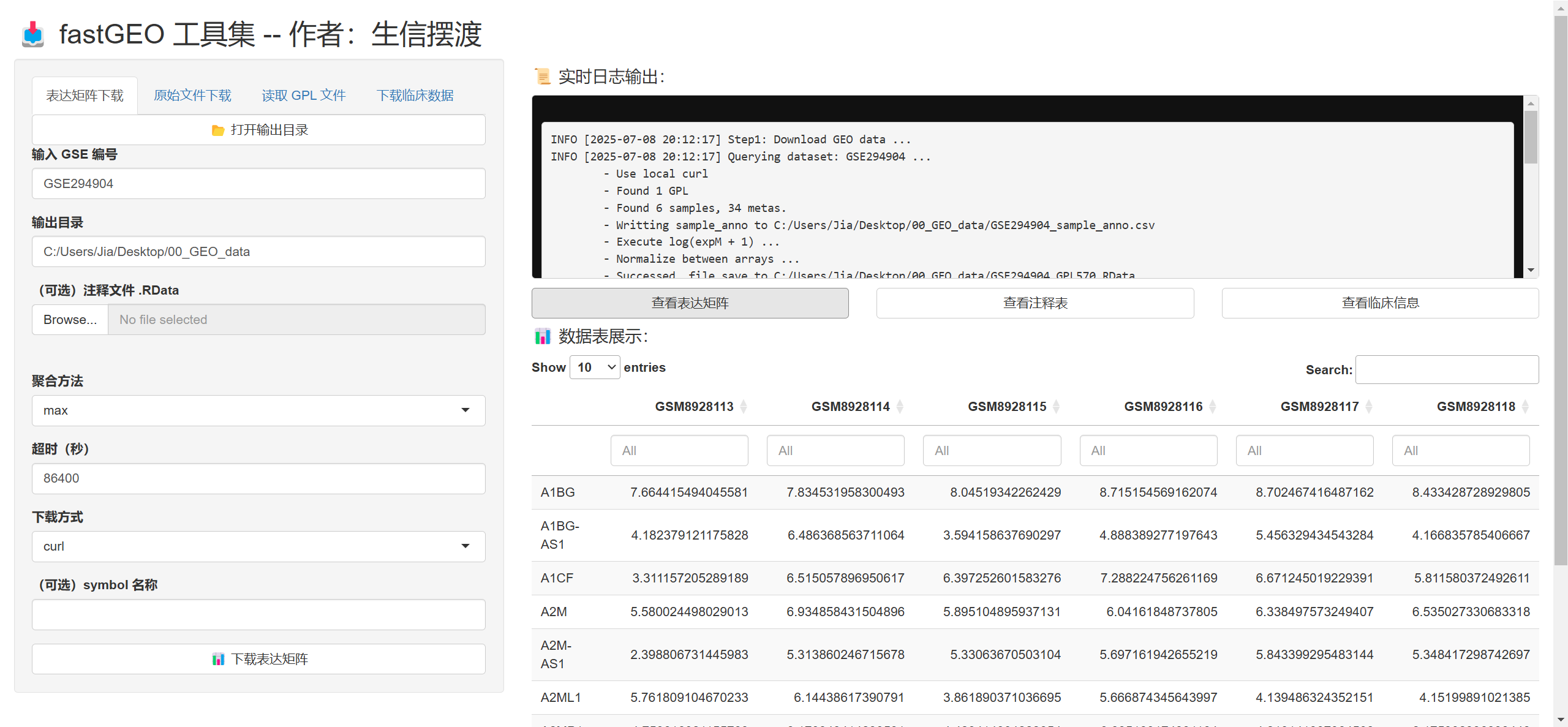
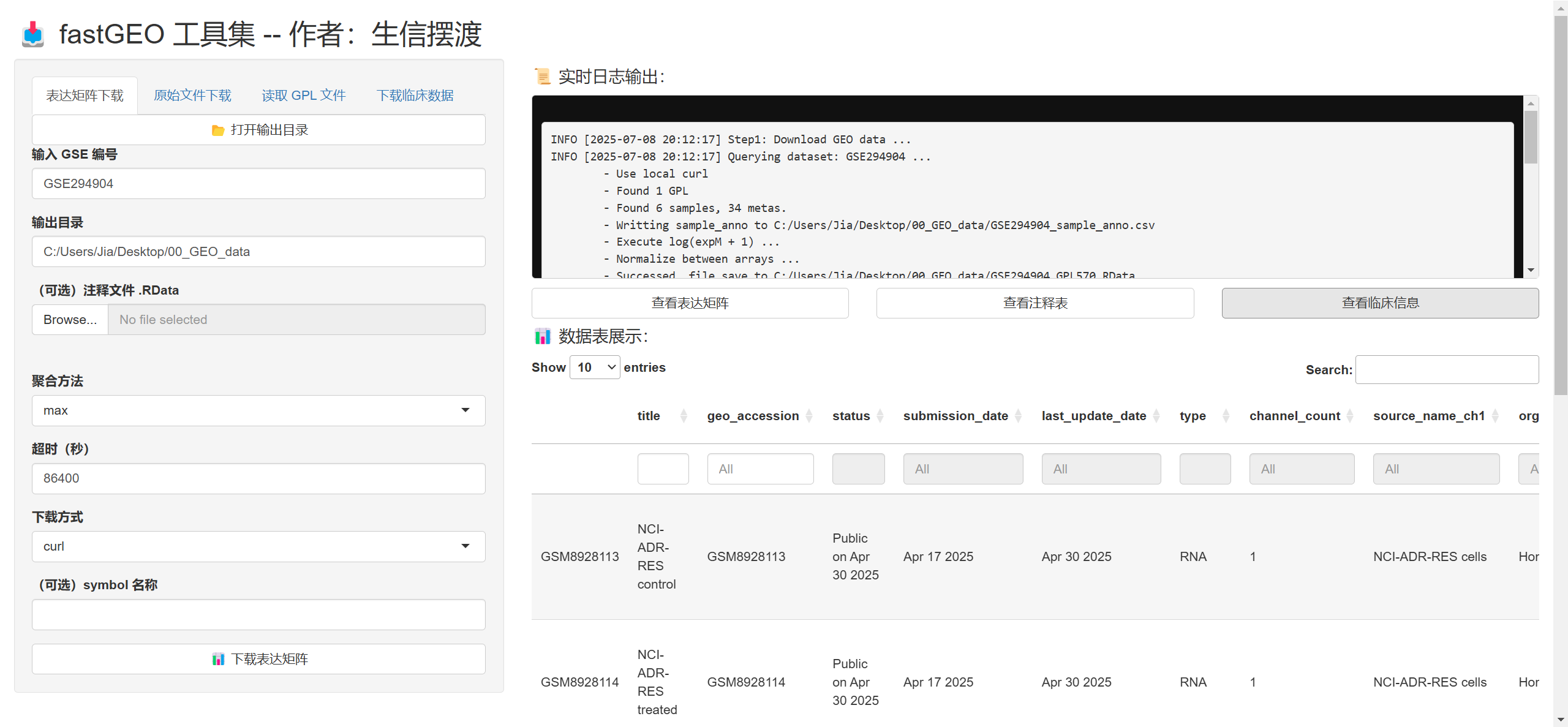
下游分析
get_GEO_group
- 根据样本信息提取并整理表达谱和分组信息
- 获取表达谱和分组信息后就可以做一系列的分析了
- 指定作为分组依据的临床信息表的列名, 及每个分组的匹配模式和分组名称即可
- 可指定两组或多组
obj = download_GEO("GSE18105", out_dir = "test/00_GEO_data_GSE18105")
>>>>> GSE18105已处理完毕, 将直接返回结果, 设置'force = TRUE'强制重新运行!
expM = obj$expM
sample_anno = obj$sample_anno
hd(expM)
Type: data.frame Dim: 22881 × 111
GSM452552 GSM452553 GSM452554 GSM452555 GSM452556
A1BG 3.884167 3.805943 3.779392 3.961285 3.980595
A1BG-AS1 4.755635 5.021991 4.992824 5.002680 4.811076
A1CF 7.603906 8.907048 8.035191 8.603941 7.788007
A2M 10.972595 7.664711 9.703405 9.684616 9.976007
A2M-AS1 4.349903 4.127930 4.240315 4.537712 4.449675
hd(sample_anno)
Type: data.frame Dim: 111 × 39
title geo_accession status submission_date last_update_date
GSM452552 patient 100, cancer, LCM GSM452552 Public on Feb 04 2010 Sep 14 2009 Aug 28 2018
GSM452553 patient 101, cancer, LCM GSM452553 Public on Feb 04 2010 Sep 14 2009 Aug 28 2018
GSM452554 patient 102, cancer, LCM GSM452554 Public on Feb 04 2010 Sep 14 2009 Aug 28 2018
GSM452555 patient 103, cancer, LCM GSM452555 Public on Feb 04 2010 Sep 14 2009 Aug 28 2018
GSM452556 patient 104, cancer, LCM GSM452556 Public on Feb 04 2010 Sep 14 2009 Aug 28 2018
table_GEO_clinical(obj$sample_anno)
$characteristics_ch1
metastasis: metastasis metastasis: metastatic recurrence metastasis: none
26 18 67
$characteristics_ch1.1
tissue: cancer, homogenized tissue: cancer, LCM tissue: normal, homogenized
17 77 17
# 两组
group_list = get_GEO_group(obj, group_name = "characteristics_ch1.1", "cancer, homogenized" = "Cancer", "normal, homogenized" = "Normal")
group = group_list$group
table(group)
group
Cancer Normal
17 17
SID = group_list$SID
hd(SID)
Type: character Dim: 34
[1] "GSM452646" "GSM452647" "GSM452648" "GSM452649" "GSM452650"
expM = group_list$expM
hd(expM)
Type: data.frame Dim: 22881 × 34
GSM452646 GSM452647 GSM452648 GSM452649 GSM452650
A1BG 4.218375 4.102014 4.080164 4.017133 3.958630
A1BG-AS1 4.813436 4.965083 4.854558 4.685886 4.796381
A1CF 8.866299 7.978485 8.947088 9.373371 9.222967
A2M 6.513392 8.517808 11.518616 8.276835 7.645857
A2M-AS1 3.880756 3.910804 5.114558 4.081589 3.817199
sample_anno = group_list$sample_anno
hd(sample_anno)
Type: data.frame Dim: 34 × 39
title geo_accession status submission_date last_update_date
GSM452646 patient 006, cancer, homogenized GSM452646 Public on Feb 04 2010 Sep 14 2009 Aug 28 2018
GSM452647 patient 011, cancer, homogenized GSM452647 Public on Feb 04 2010 Sep 14 2009 Aug 28 2018
GSM452648 patient 024, cancer, homogenized GSM452648 Public on Feb 04 2010 Sep 14 2009 Aug 28 2018
GSM452649 patient 027, cancer, homogenized GSM452649 Public on Feb 04 2010 Sep 14 2009 Aug 28 2018
GSM452650 patient 028, cancer, homogenized GSM452650 Public on Feb 04 2010 Sep 14 2009 Aug 28 2018
# 多组
group_list = get_GEO_group(obj, group_name = "characteristics_ch1.1",
"cancer, homogenized" = "Cancer",
"normal, homogenized" = "Normal",
"cancer, LCM" = "Cancer_LCM"
)
table(group_list$group)
Cancer Cancer_LCM Normal
17 77 17
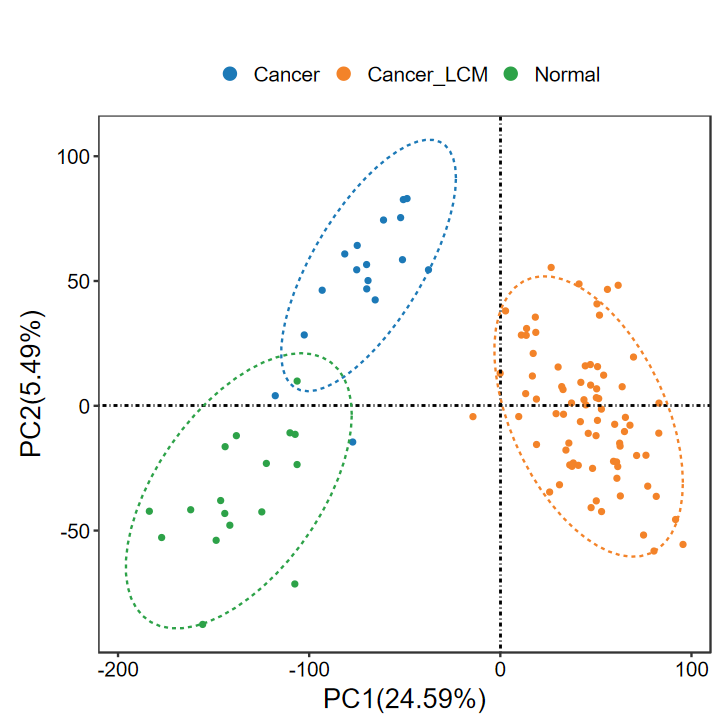
run_PCA
- 执行PCA分析并绘图
- 默认添加椭圆、默认不添加样本标签
- 可输出图片(PDF/PNG), 返回ggplot对象, 可自行修改
group = group_list$group
expM = group_list$expM
SID = group_list$SID
table(group)
group
Cancer Cancer_LCM Normal
17 77 17
set_image(6, 6)
run_PCA(expM, group, out_name = "./test/00_GEO_data_GSE18105/01_PCA")
# 修改属性
run_PCA(expM, group,
title = "PCA Plot",
legend.title = "Group",
text.size = 30,
pt.size = 5,
key.size = 5,
color = c("red", "green", "blue")
)
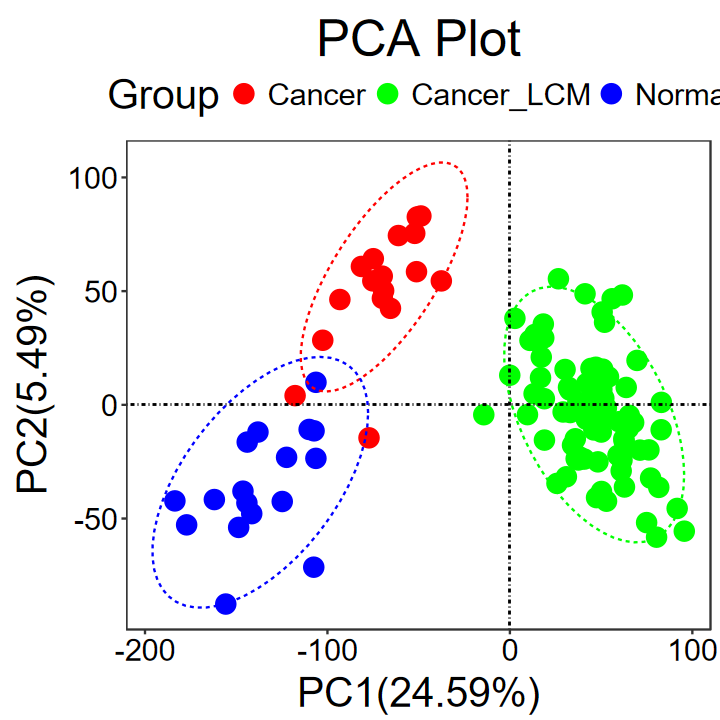
- 添加label, 适合样本较少的情况, 可以更清晰地展示离群样本名
- 这里每组只提取前5个样本展示
SID2 = unlist(lapply(group_list$SID_list, function(x) x[1:5]))
group2 = group[match(SID2, SID)]
expM2 = expM[, SID2]
table(group2)
group2
Cancer Cancer_LCM Normal
5 5 5
run_PCA(expM2, group2, label = TRUE)
Warning message in geom_text_repel(aes(label = Sample), linewidth = ifelse(is.null(label.size), :
"[1m[22mIgnoring unknown parameters: `linewidth`"
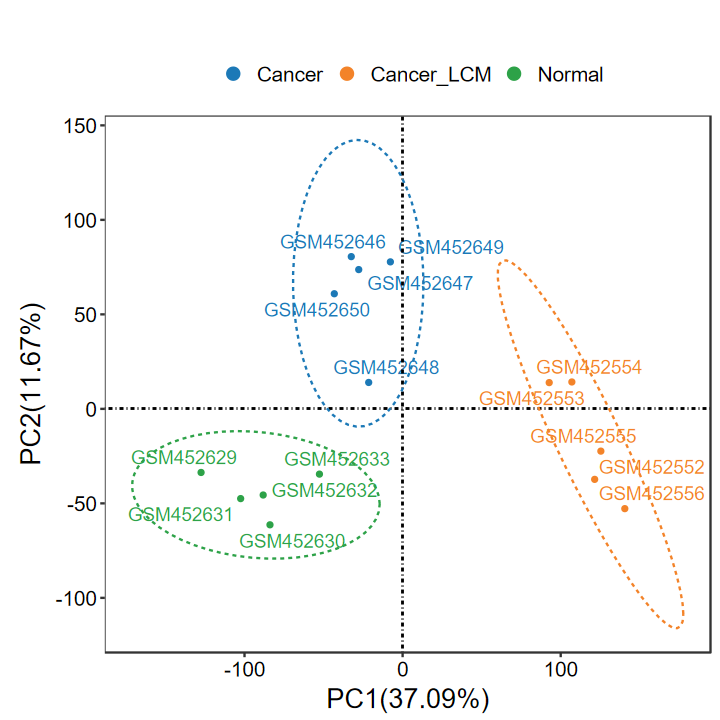
run_DEG_limma
- 快速进行limma差异分析, 适合标准化的芯片数据或高通量数据
- 可设置阈值, 默认 |log2FC| > 1, Padj < 0.05
- 可设置不使用P值矫正
- 可添加基因label, 可自定义基因
DEG_tb = run_DEG_limma(expM, group, Case = "Cancer", Control = "Normal", out_name = "./test/00_GEO_data_GSE18105/02_DEG")
Warning message:
"Parameter 'Case' is deprecated since fastGEO 1.8.0, please use 'case' instead!"
Warning message:
"Parameter 'Control' is deprecated since fastGEO 1.8.0, please use 'control' instead!"
Number of Up regulated genes: 1006/2102(47.86%)
Number of Down regulated genes: 1096/2102(52.14%)
Number of Not Change genes: 20779/22881
head(DEG_tb)
| logFC | AveExpr | t | P.Value | adj.P.Val | B | DEG | |
|---|---|---|---|---|---|---|---|
| <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <dbl> | <chr> | |
| FOXQ1 | 5.716354 | 8.777030 | 13.702008 | 2.117724e-15 | 8.075941e-12 | 24.97073 | Up |
| CLDN1 | 4.640807 | 8.502369 | 19.330354 | 6.543012e-20 | 1.497107e-15 | 34.74020 | Up |
| LGR5 | 4.485429 | 7.354803 | 10.223319 | 6.637827e-12 | 1.421583e-09 | 17.14899 | Up |
| KRT23 | 4.445303 | 6.613692 | 8.937107 | 1.917386e-10 | 1.589992e-08 | 13.84479 | Up |
| DPEP1 | 4.260363 | 8.240490 | 10.122520 | 8.572521e-12 | 1.662270e-09 | 16.89826 | Up |
| CEMIP | 4.147864 | 8.048877 | 12.218476 | 5.487202e-14 | 5.879765e-11 | 21.82719 | Up |
# 最低阈值
DEG_tb2 = run_DEG_limma(expM, group, Case = "Cancer", Control = "Normal", log2FC = 0.5, padj = FALSE, out_name = "./test/00_GEO_data_GSE18105/03_DEG")
Warning message:
"Parameter 'Case' is deprecated since fastGEO 1.8.0, please use 'case' instead!"
Warning message:
"Parameter 'Control' is deprecated since fastGEO 1.8.0, please use 'control' instead!"
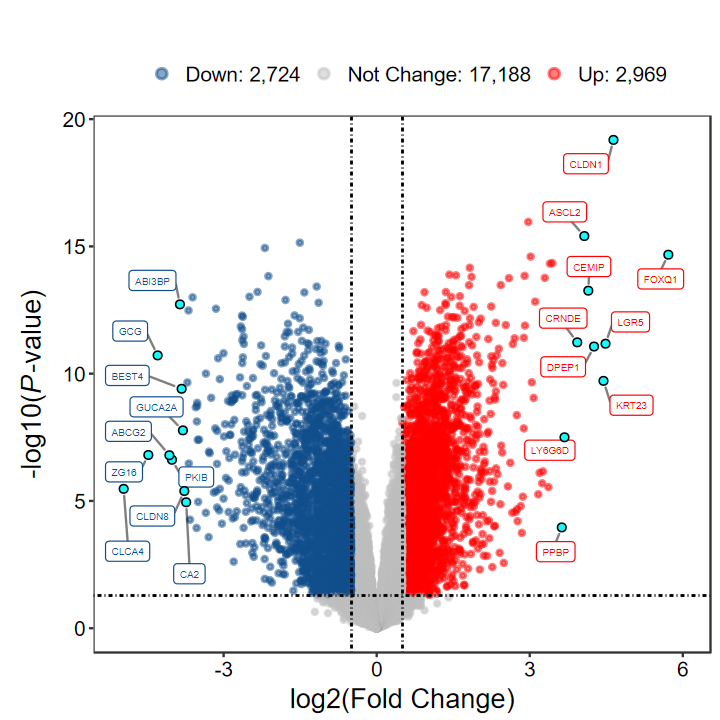
Number of Up regulated genes: 2969/5693(52.15%)
Number of Down regulated genes: 2724/5693(47.85%)
Number of Not Change genes: 17188/22881
plot_volcano_limma
- 根据差异表格绘制火山图
group_list = get_GEO_group(obj, group_name = "characteristics_ch1.1",
"cancer, homogenized" = "Cancer",
"normal, homogenized" = "Normal",
"cancer, LCM" = "Cancer_LCM"
)
table(group_list$group)
Cancer Cancer_LCM Normal
17 77 17
group = group_list$group
expM = group_list$expM
SID = group_list$SID
table(group)
group
Cancer Cancer_LCM Normal
17 77 17
DEG_tb = run_DEG_limma(expM, group, Case = "Cancer", Control = "Normal", out_name = "./test/00_GEO_data_GSE18105/02_DEG")
Warning message:
"Parameter 'Case' is deprecated since fastGEO 1.8.0, please use 'case' instead!"
Warning message:
"Parameter 'Control' is deprecated since fastGEO 1.8.0, please use 'control' instead!"
Number of Up regulated genes: 1006/2102(47.86%)
Number of Down regulated genes: 1096/2102(52.14%)
Number of Not Change genes: 20779/22881
plot_volcano_limma(DEG_tb, out_name = "./test/00_GEO_data_GSE18105/04_volcano")
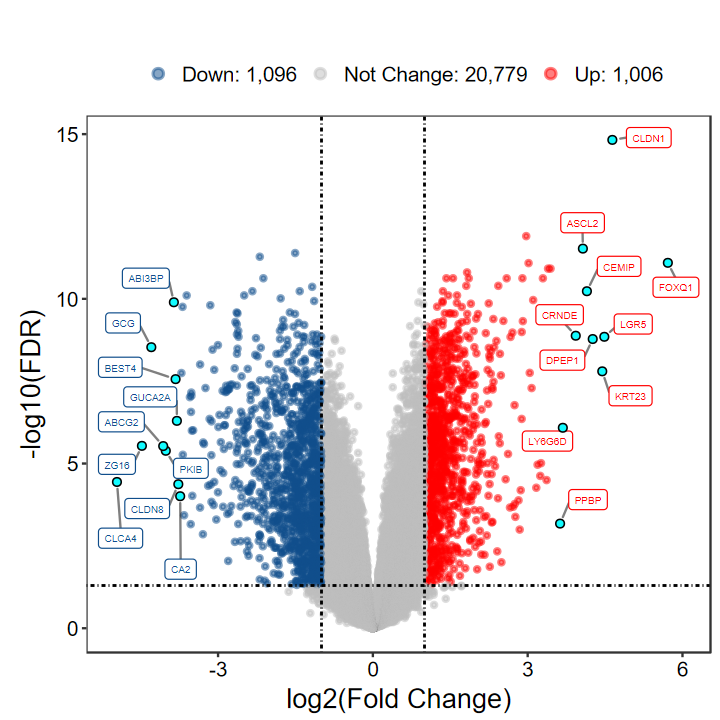
# 显示阈值
plot_volcano_limma(DEG_tb, caption = TRUE)
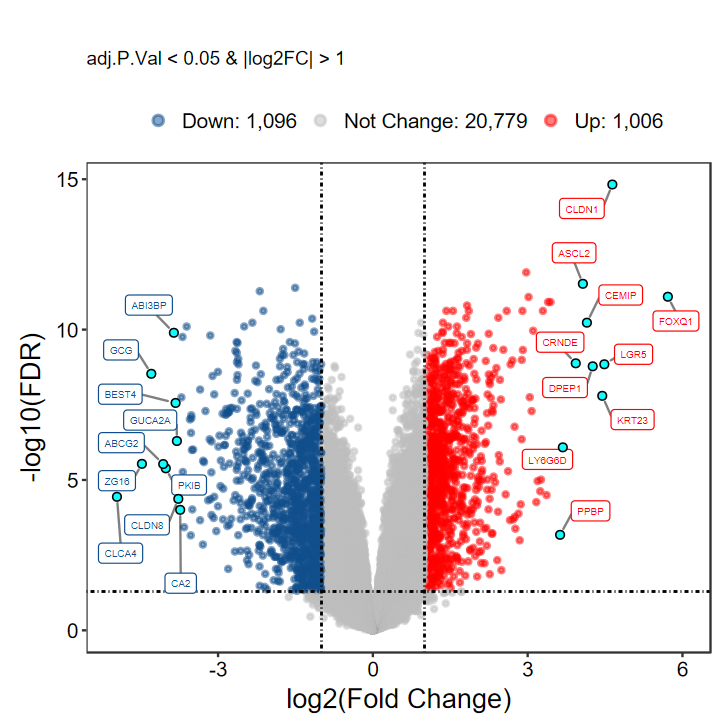
# 选择 top logFC 前10的基因进行显示
plot_volcano_limma(DEG_tb, caption = TRUE, method = "logFC")
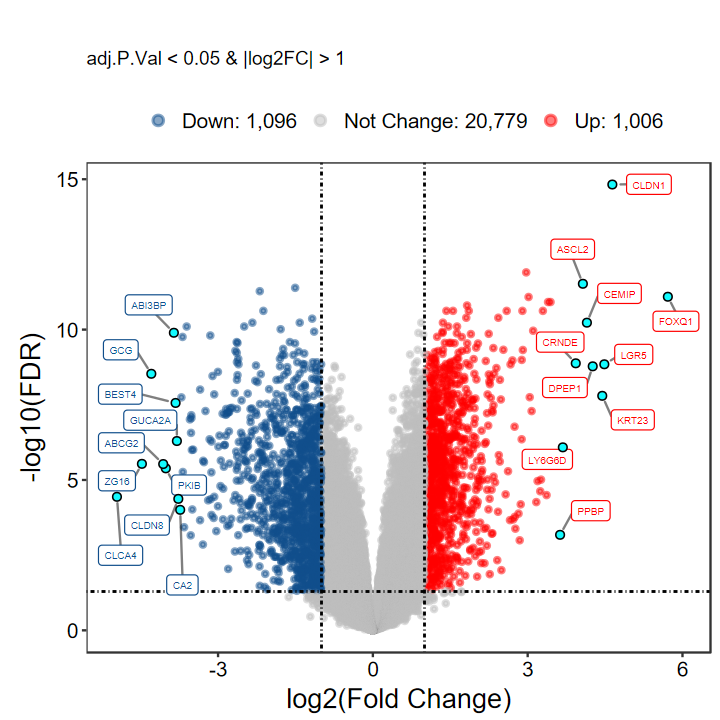
# 不显示基因名
plot_volcano_limma(DEG_tb, caption = TRUE, label = FALSE)
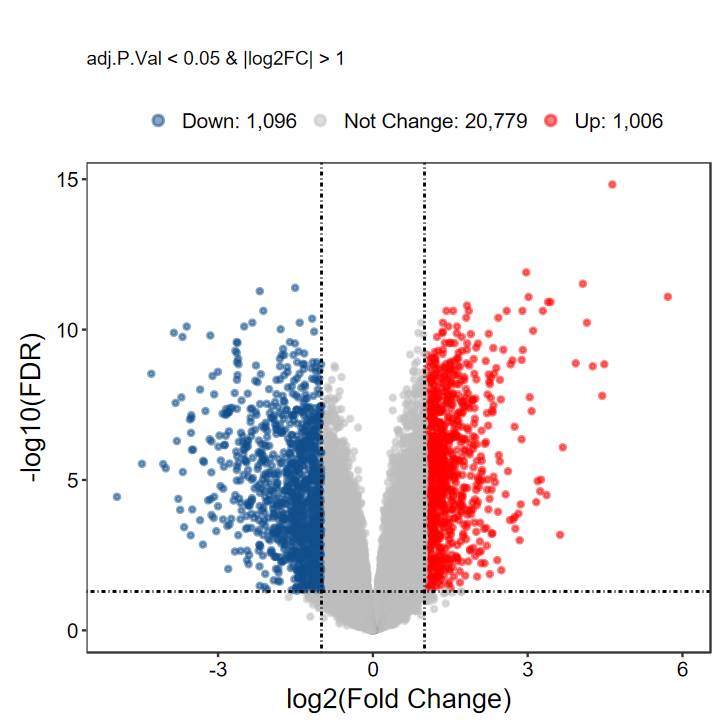
# 自定义label基因, 每组随机挑选5个
set.seed(1234)
gene_slect = as.character(aggregate(rownames(DEG_tb), by = list(DEG_tb$DEG), function(x) sample(x, 5))[, 2])
gene_slect
- 'CPM'
- 'STX16'
- 'TTC27'
- 'FFAR4'
- 'RP11-31K23.2'
- 'MCM2'
- 'GNA11'
- 'PIK3IP1'
- 'RFC3'
- 'CAPN9'
- 'TFPI'
- 'ASCC3'
- 'DMXL2'
- 'GK5'
- 'SLC2A1'
plot_volcano_limma(DEG_tb, caption = TRUE, label = gene_slect)
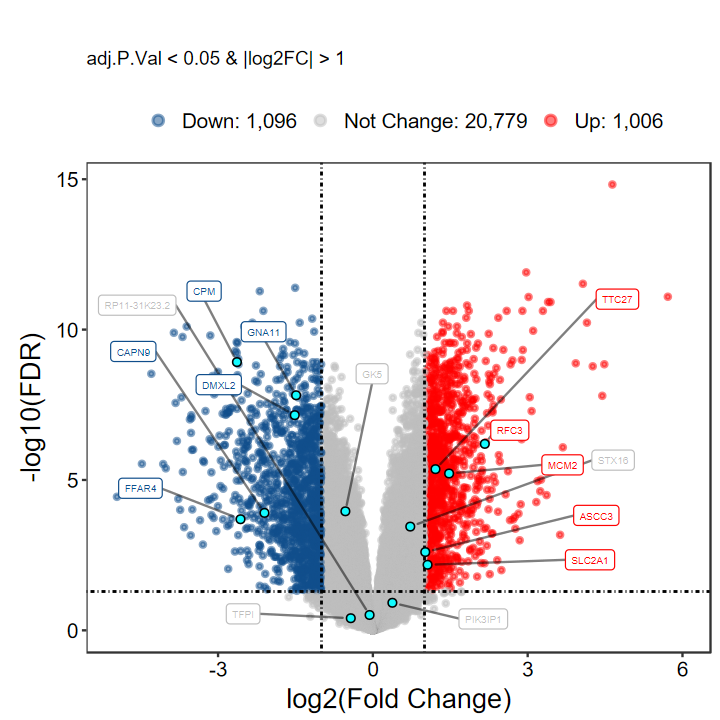
- 如果差异分析的阈值发生改变, 这里也要进行一样的设置
# 最低阈值
DEG_tb2 = run_DEG_limma(expM, group, Case = "Cancer", Control = "Normal", log2FC = 0.5, padj = FALSE, out_name = "./test/00_GEO_data_GSE18105/03_DEG")
Warning message:
"Parameter 'Case' is deprecated since fastGEO 1.8.0, please use 'case' instead!"
Warning message:
"Parameter 'Control' is deprecated since fastGEO 1.8.0, please use 'control' instead!"
Number of Up regulated genes: 2969/5693(52.15%)
Number of Down regulated genes: 2724/5693(47.85%)
Number of Not Change genes: 17188/22881
plot_volcano_limma(DEG_tb2, log2FC = 0.5, padj = FALSE)
plot_heatmap_DEG
- 根据差异分析结果和表达谱绘制差异基因热图
# 仅支持输入两种分组
group2 = group[group != "Cancer_LCM"]
expM2 = expM[, group != "Cancer_LCM"]
table(group2)
group2
Cancer Normal
17 17
plot_heatmap_DEG(expM2, DEG_tb, group2, out_name = "./test/00_GEO_data_GSE18105/04_heatmap")
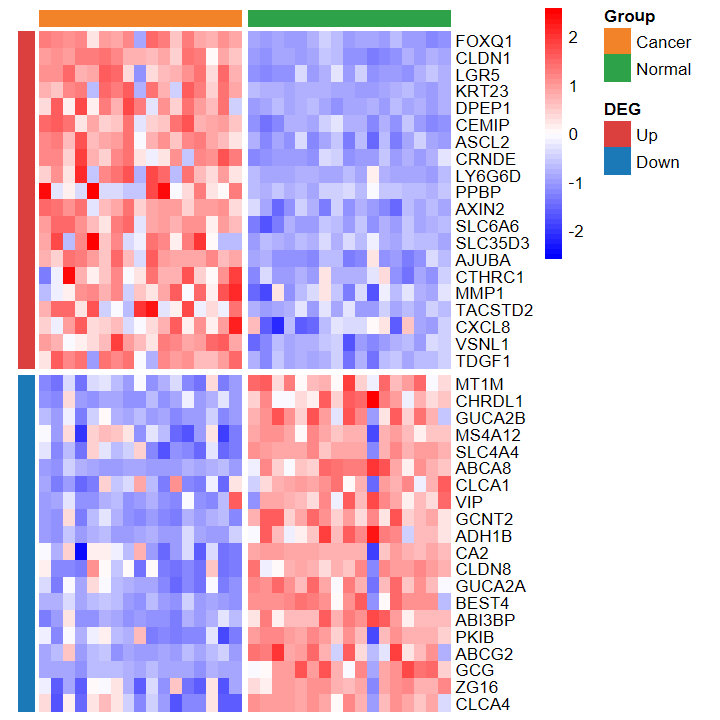
set_image(7, 10)
plot_heatmap_DEG(expM2, DEG_tb, group2, ntop = 30, w = 15)
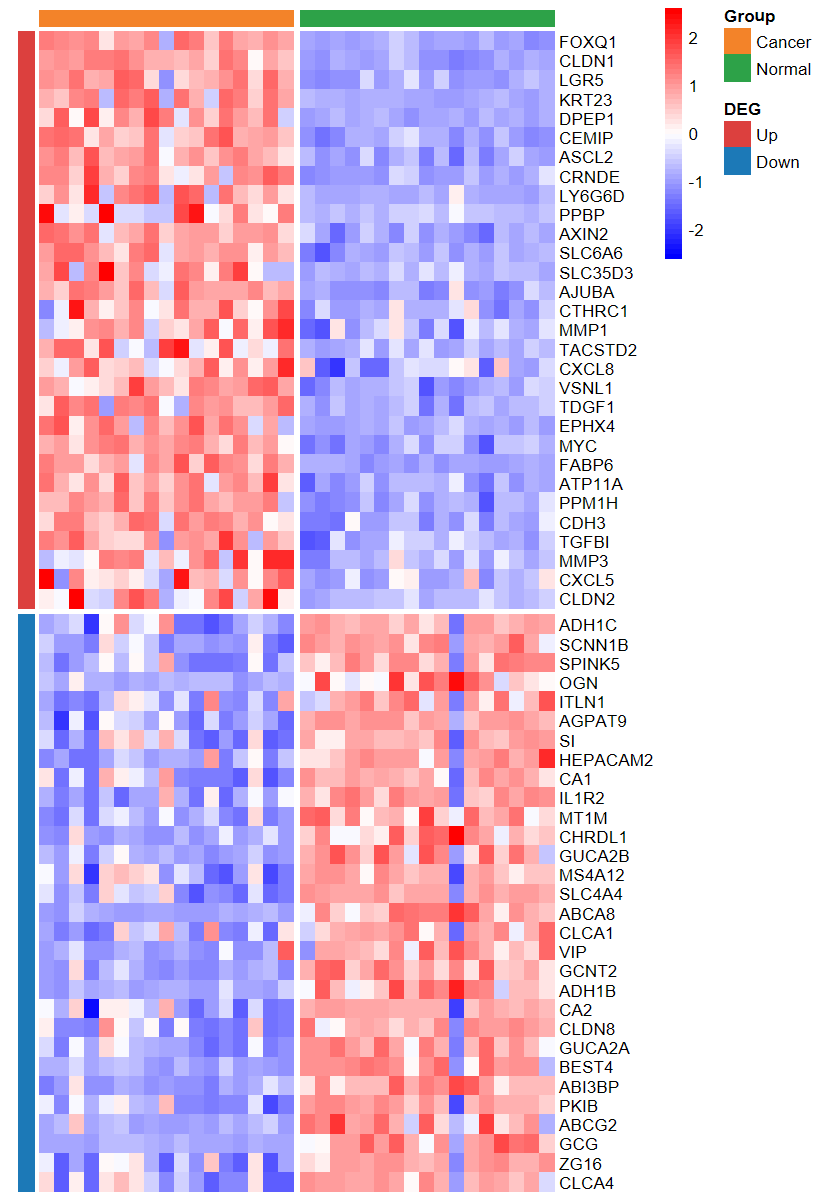
plot_boxplot
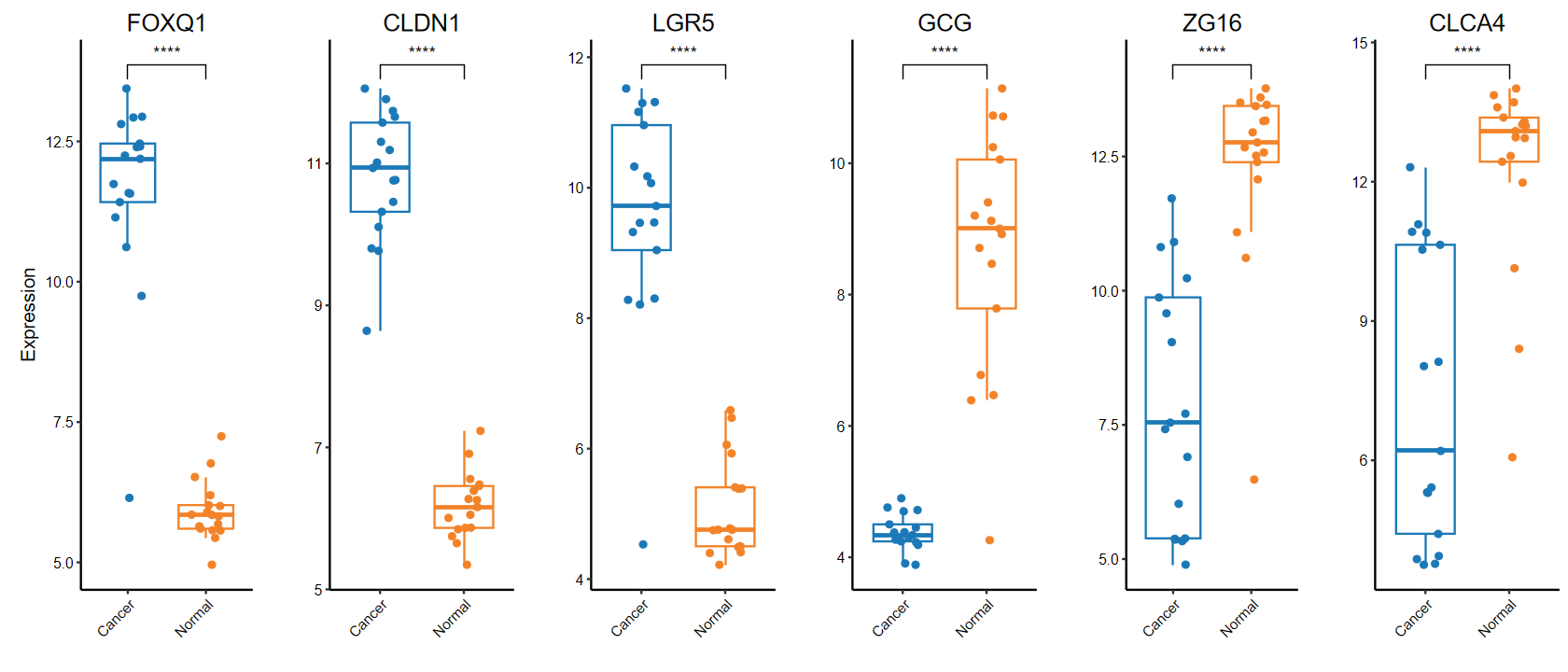
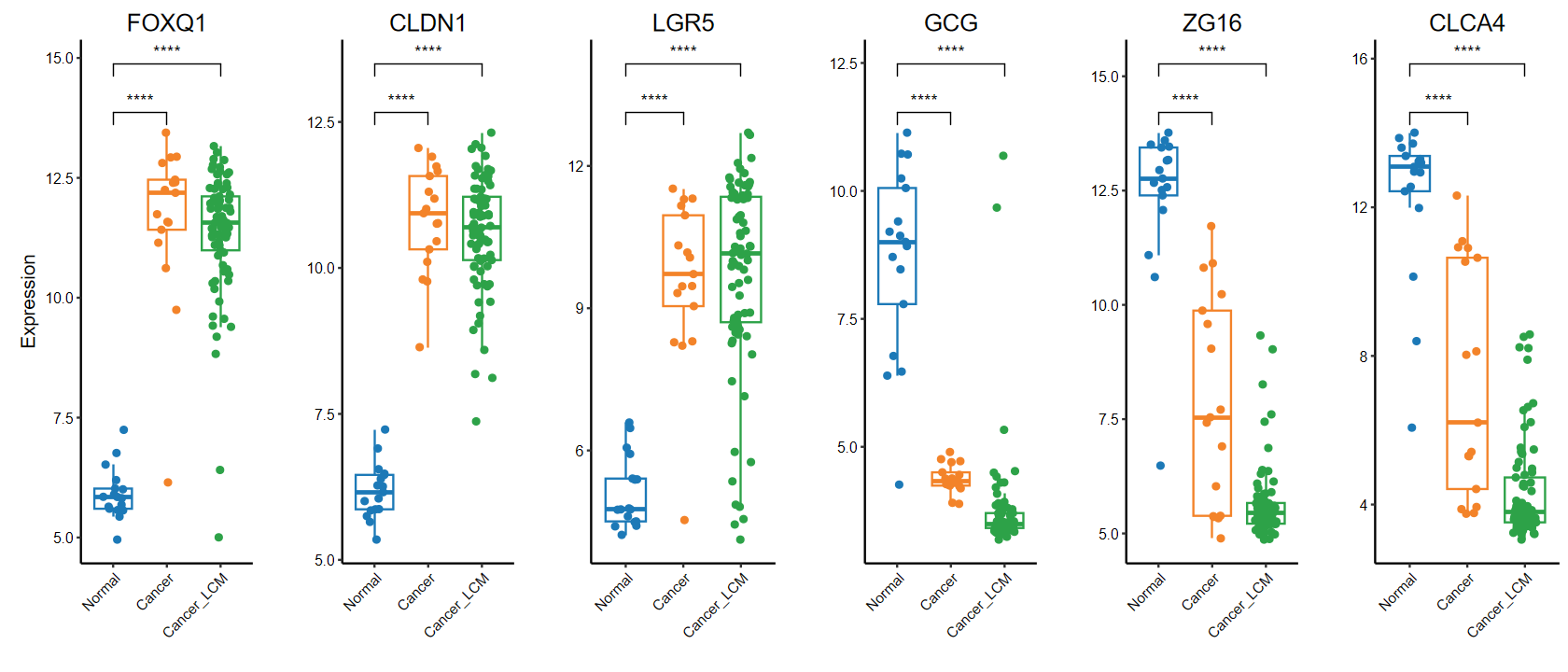
gene_select = c(head(rownames(DEG_tb), 3), tail(rownames(DEG_tb), 3))
gene_select
- 'FOXQ1'
- 'CLDN1'
- 'LGR5'
- 'GCG'
- 'ZG16'
- 'CLCA4'
set_image(14, 6)
plot_boxplot(expM2, group2,
genes = gene_select,
out_name = "./test/00_GEO_data_GSE18105/06_boxplot", w = 10, h = 5)
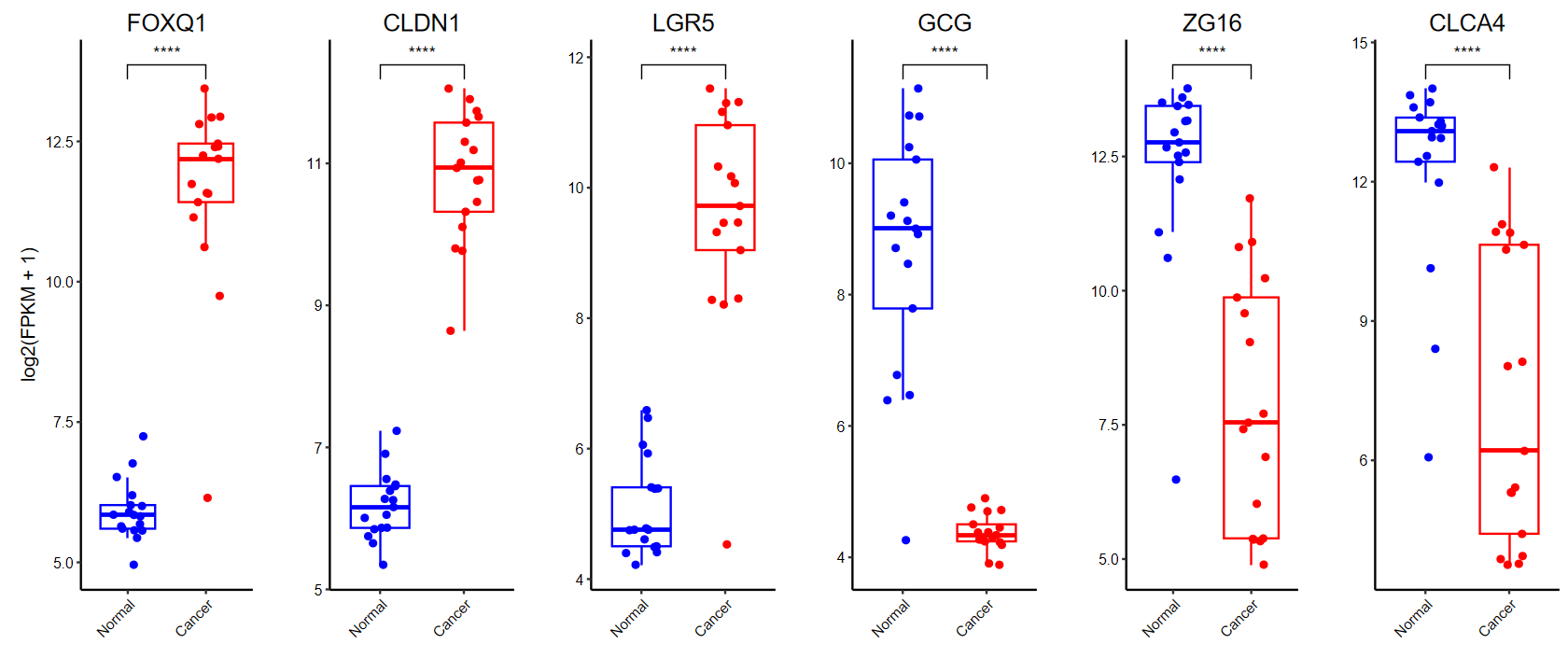
set_image(14, 6)
plot_boxplot(expM2, group2,
genes = gene_select,
breaks = c("Normal", "Cancer"), # 修改x轴顺序
ylab = "log2(FPKM + 1)", # 修改y轴标题
color = c("blue", "red"), # 修改颜色
w = 10, h = 5)
# 多组
set_image(14, 6)
plot_boxplot(expM, group,
breaks = c("Normal", "Cancer", "Cancer_LCM"), # 修改x轴顺序
comparisons = list(c("Cancer", "Normal"), c("Cancer_LCM", "Normal")), # 手动指定分组
genes = gene_select)
run_DEG_deseq2
- 快速使用DESeq2进行差异分析, 适合count数据
- 有count数据优先使用DESeq2
load2("./test/TCGA/00_TCGA_data.RData")
Loading objects:
expM
expM_FPKM
group
sample_anno
hd(expM)
Type: data.frame Dim: 59427 × 562
TCGA-60-2712-01A-01R-0851-07 TCGA-56-7221-01A-11R-2045-07 TCGA-21-A5DI-01A-31R-A26W-07 TCGA-43-7657-01A-31R-2125-07
5_8S_rRNA 0 0 0 0
5S_rRNA 0 0 0 4
7SK 0 3 0 0
A1BG 7 1 0 6
A1BG-AS1 81 34 31 41
TCGA-94-7033-01A-11R-1949-07
5_8S_rRNA 0
5S_rRNA 4
7SK 0
A1BG 5
A1BG-AS1 24
table(group)
group
LUAD Normal
511 51
sum(group == "Normal")
51
# 取子集测试
set.seed(1234)
SID_tumor = sample(colnames(expM)[group == "LUAD"], sum(group == "Normal"))
expM2 = expM[, c(SID_tumor, colnames(expM)[group == "Normal"])]
group2 = group[match(colnames(expM2), colnames(expM))]
table(group2)
group2
LUAD Normal
51 51
DEG_tb_count = run_DEG_deseq2(expM2, group2, Case = "LUAD", Control = "Normal", out_name = "./test/TCGA/TCGA_LUAD_DEG")
estimating size factors
estimating dispersions
gene-wise dispersion estimates
mean-dispersion relationship
final dispersion estimates
fitting model and testing
-- replacing outliers and refitting for 3453 genes
-- DESeq argument 'minReplicatesForReplace' = 7
-- original counts are preserved in counts(dds)
estimating dispersions
fitting model and testing
Number of Up regulated genes: 6032
Number of Down regulated genes: 11035
Number of Not Change regulated genes: 37154
count_to_exp
- 用于将count矩阵转为标准化的表达谱, 可用于绘制PCA、热图和箱线图等
- 不依赖于基因长度, 省去计算TPM/FPKM的麻烦
- 使用 DESeq2 的 VST 标准化方法
expM_vst = count_to_exp(expM2)
hd(expM_vst)
Type: data.frame Dim: 59427 × 102
TCGA-58-A46K-01A-11R-A24H-07 TCGA-68-A59J-01A-21R-A26W-07 TCGA-66-2753-01A-01R-0980-07 TCGA-77-8136-01A-11R-2247-07
5_8S_rRNA 1.885298 1.885298 1.885298 1.885298
5S_rRNA 3.184227 1.885298 2.586642 1.885298
7SK 1.885298 1.885298 1.885298 1.885298
A1BG 3.452268 3.881663 3.250346 4.269259
A1BG-AS1 5.402377 5.804480 5.639623 6.755954
TCGA-46-3765-01A-01R-0980-07
5_8S_rRNA 1.885298
5S_rRNA 1.885298
7SK 1.885298
A1BG 3.959024
A1BG-AS1 5.297581
expM_FPKM2 = expM_FPKM[, colnames(expM2)]
hd(expM_FPKM2)
Type: data.frame Dim: 59427 × 102
TCGA-58-A46K-01A-11R-A24H-07 TCGA-68-A59J-01A-21R-A26W-07 TCGA-66-2753-01A-01R-0980-07 TCGA-77-8136-01A-11R-2247-07
5_8S_rRNA 0.0000000 0.0000000 0.00000000 0.0000000
5S_rRNA 1.7187452 0.0000000 0.63133714 0.0000000
7SK 0.0000000 0.0000000 0.00000000 0.0000000
A1BG 0.1257833 0.1797658 0.08161232 0.2777468
A1BG-AS1 0.8414901 0.9455328 0.87018715 1.5912964
TCGA-46-3765-01A-01R-0980-07
5_8S_rRNA 0.0000000
5S_rRNA 0.0000000
7SK 0.0000000
A1BG 0.1615653
A1BG-AS1 0.5865966
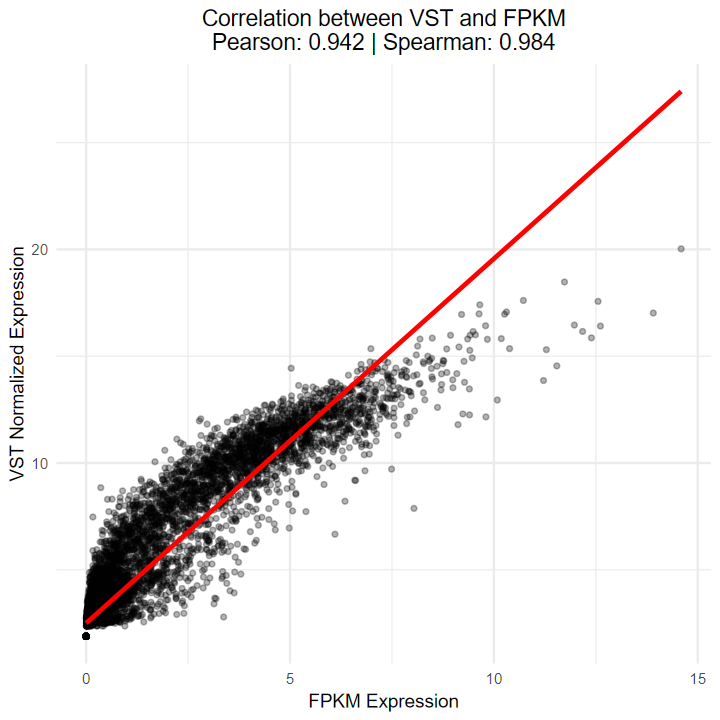
- 测试vst算法与TCGA官网提供的FPKM的相关性
- 结果显示Sperman相关性达到0.984, 说明相关性极高, 可以等价
# 加载必需的包(确保版本兼容)
if (!require("dplyr", quietly = TRUE)) {
install.packages("dplyr")
library(dplyr)
}
if (!require("ggplot2", quietly = TRUE)) {
install.packages("ggplot2")
library(ggplot2)
}
# 确保两个矩阵的基因和样本匹配
common_genes <- intersect(rownames(expM_vst), rownames(expM_FPKM2))
common_samples <- intersect(colnames(expM_vst), colnames(expM_FPKM2))
expM_vst_filtered <- expM_vst[common_genes, common_samples]
expM_FPKM_filtered <- expM_FPKM2[common_genes, common_samples]
# 转换为长格式(不使用rownames_to_column)
# 处理VST数据:手动添加基因名列
vst_df <- as.data.frame(expM_vst_filtered)
vst_df$gene <- rownames(vst_df) # 手动将行名转为gene列
vst_long <- tidyr::pivot_longer(vst_df, cols = -gene,
names_to = "sample", values_to = "vst_value")
# 处理FPKM数据:手动添加基因名列
fpkm_df <- as.data.frame(expM_FPKM_filtered)
fpkm_df$gene <- rownames(fpkm_df) # 手动将行名转为gene列
fpkm_long <- tidyr::pivot_longer(fpkm_df, cols = -gene,
names_to = "sample", values_to = "fpkm_value")
# 合并数据
combined_data <- inner_join(vst_long, fpkm_long, by = c("gene", "sample"))
# 计算相关性
cor_pearson <- cor(combined_data$vst_value, combined_data$fpkm_value, method = "pearson")
cor_spearman <- cor(combined_data$vst_value, combined_data$fpkm_value, method = "spearman")
hd(combined_data)
Type: tbl_df Dim: 6061554 × 4
gene sample vst_value fpkm_value
1 5_8S_rRNA TCGA-58-A46K-01A-11R-A24H-07 1.885298 0
2 5_8S_rRNA TCGA-68-A59J-01A-21R-A26W-07 1.885298 0
3 5_8S_rRNA TCGA-66-2753-01A-01R-0980-07 1.885298 0
4 5_8S_rRNA TCGA-77-8136-01A-11R-2247-07 1.885298 0
5 5_8S_rRNA TCGA-46-3765-01A-01R-0980-07 1.885298 0
# 绘制散点图
set_image(6, 6)
set.seed(1234)
combined_data = data.frame(combined_data)
combined_data2 = combined_data[sample(1:nrow(combined_data), 10000), ]
# 计算相关性
cor_pearson <- cor(combined_data2$vst_value, combined_data2$fpkm_value, method = "pearson")
cor_spearman <- cor(combined_data2$vst_value, combined_data2$fpkm_value, method = "spearman")
ggplot(combined_data2, aes(x = fpkm_value, y = vst_value)) +
geom_point(alpha = 0.3, size = 1) +
geom_smooth(method = "lm", color = "red", se = FALSE) +
labs(
x = "FPKM Expression",
y = "VST Normalized Expression",
title = paste0("Correlation between VST and FPKM\n",
"Pearson: ", round(cor_pearson, 3), " | ",
"Spearman: ", round(cor_spearman, 3))
) +
theme_minimal() +
theme(plot.title = element_text(hjust = 0.5))
[1m[22m`geom_smooth()` using formula = 'y ~ x'
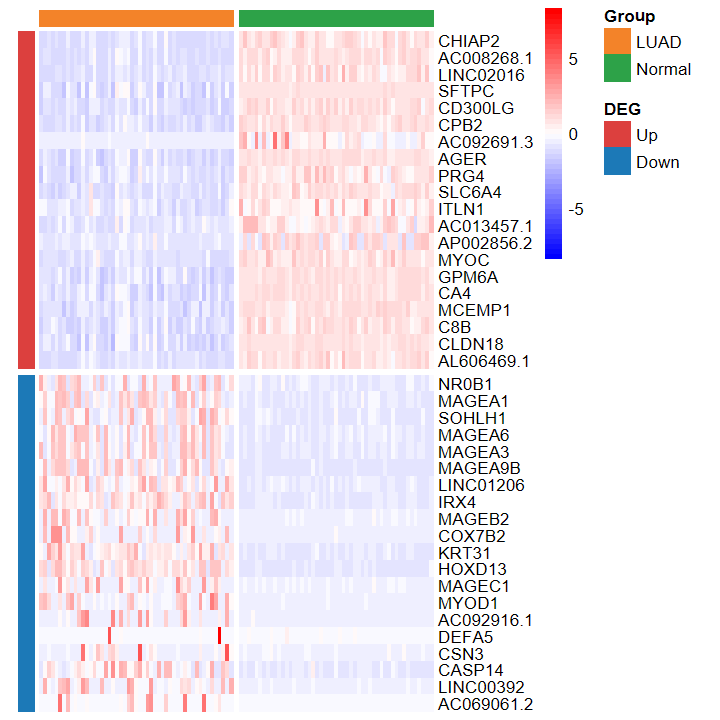
- 绘制的热图也看不出差异
plot_heatmap_DEG(expM_vst, DEG_tb_count, group2)
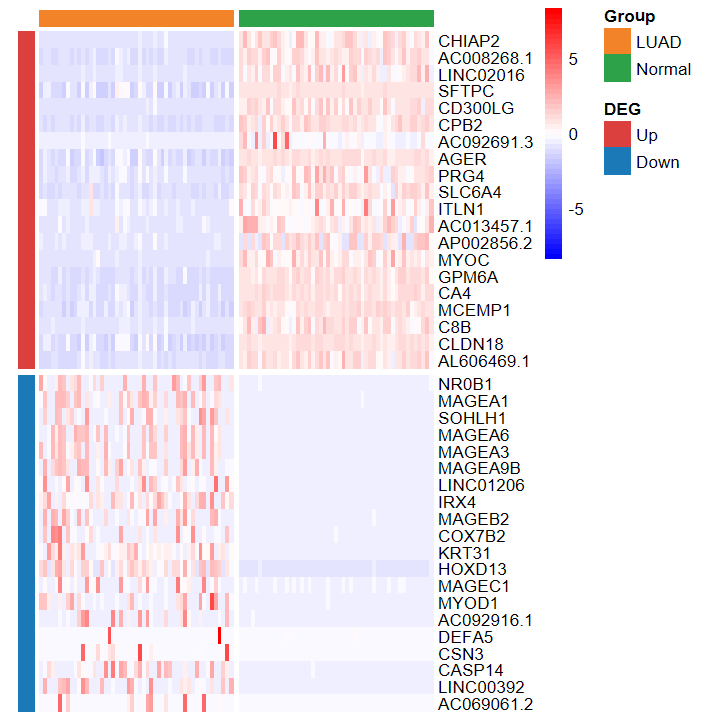
plot_heatmap_DEG(expM_FPKM2, DEG_tb_count, group2)
run_ComBat
- 去除多个数据集之间的批次效应, 并绘制去批次前后的PCA图
# 下载处理 GSE108109 芯片数据
obj = download_GEO("GSE108109", out_dir = "./test/run_ComBat")
>>>>> 识别到本地注释文件, 将优先使用!
>>>>> 访问数据集: GSE108109...
>>>>> 下载尝试次数: 1 ... FALSE
下载成功!
>>>>> 处理测序平台: GPL19983
>>>>> 识别到111个样本, 34个注释信息
>>>>> 输出样本注释表至: ./test/run_ComBat/GSE108109_GPL19983_sample_anno.csv
>>>>> 执行样本间矫正...
>>>>> 处理成功文件保存至: ./test/run_ComBat/GSE108109_GPL19983_raw.RData
>>>>> 执行探针-基因注释, 平台: GPL19983 ...
>>>>> 使用R包内置的注释文件...
>>>>> 去除重复的基因注释, 使用方法: max...
>>>>> 处理结束!
obj_list = get_GEO_group(obj, group_name = "source_name_ch1",
"Membranous Nephropathy" = "MN",
"Living donor" = "Control"
)
expM_GSE108109 = obj_list$expM
group_GSE108109 = obj_list$group
#下载处理 GSE216841 高通量
sample_anno = get_GEO_pheno("GSE216841")
>>>>> 访问数据集: GSE216841...
>>>>> 下载尝试次数: 1 ... FALSE
下载成功!
>>>>> 处理测序平台: GPL20301
>>>>> 识别到34个样本, 40个注释信息
>>>>> 输出样本注释表至: 00_GEO_data/GSE216841_GPL20301_sample_anno.csv
>>>>> 未识别到表达谱, 提供的数据集可能是高通量测序, 请确认!
https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE216841
>>>>> 高通量数据的表达谱请自行从补充文件下载处理!
>>>>> 将仅返回样本注释表!
hd(sample_anno)
Type: data.frame Dim: 34 × 40
title geo_accession status submission_date last_update_date
GSM6696604 Normal control 1 GSM6696604 Public on Jan 25 2023 Oct 28 2022 Jan 25 2023
GSM6696605 Normal control 2 GSM6696605 Public on Jan 25 2023 Oct 28 2022 Jan 25 2023
GSM6696606 Normal control 3 GSM6696606 Public on Jan 25 2023 Oct 28 2022 Jan 25 2023
GSM6696607 Normal control 4 GSM6696607 Public on Jan 25 2023 Oct 28 2022 Jan 25 2023
GSM6696608 Normal control 5 GSM6696608 Public on Jan 25 2023 Oct 28 2022 Jan 25 2023
expM_raw = read.faster("./test/run_ComBat/GSE216841_MNd_ncounts_annot.txt.gz")
expM_raw = col2rownames(expM_raw)
hd(expM_raw)
Type: data.frame Dim: 20321 × 37
ensembl_gene_id hgnc_GRCh38p12 description
ENSG00000000003 ENSG00000000003 TSPAN6 tetraspanin 6 [Source:HGNC Symbol;Acc:HGNC:11858]
ENSG00000000005 ENSG00000000005 TNMD tenomodulin [Source:HGNC Symbol;Acc:HGNC:17757]
ENSG00000000419 ENSG00000000419 DPM1 dolichyl-phosphate mannosyltransferase subunit 1, catalytic [Source:HGNC Symbol;Acc:HGNC:3005]
ENSG00000000457 ENSG00000000457 SCYL3 SCY1 like pseudokinase 3 [Source:HGNC Symbol;Acc:HGNC:19285]
ENSG00000000460 ENSG00000000460 C1orf112 chromosome 1 open reading frame 112 [Source:HGNC Symbol;Acc:HGNC:25565]
N_CTRL1 N_CTRL2
ENSG00000000003 92.874228 309.359955
ENSG00000000005 1.020596 1.041616
ENSG00000000419 255.148977 1.041616
ENSG00000000457 373.538103 887.456841
ENSG00000000460 110.224358 289.569251
countM = expM_raw[, -c(1:3)]
name_list = c("N_CTRL" = "Normal control", "MEMBR" = "Idiopathic membranous nephropathy", "MCD" = "Minimal change disease")
new_name = paste(name_list[paste0(gsub("[0-9]", "", colnames(countM)))], gsub("[A-Z_]", "", colnames(countM)))
colnames(countM) = sample_anno$geo_accession[match(new_name, sample_anno$title)]
countM = ceiling(countM)
countM = data.frame(SYMBOL = expM_raw$hgnc_GRCh38p12, countM)
countM = aggregate(. ~ SYMBOL, data = countM, function(x) x[which.max(x)])
countM = col2rownames(countM)
expM = count_to_exp(countM)
converting counts to integer mode
group = subString(sample_anno[colnames(countM), "title"], -1, " ", rev = TRUE, collapse = " ")
table(group)
group
Idiopathic membranous nephropathy Minimal change disease Normal control
12 14 8
SID_MN = colnames(expM)[group == "Idiopathic membranous nephropathy"]
SID_Control = colnames(expM)[group == "Normal control"]
expM_GSE216841 = expM[, c(SID_MN, SID_Control)]
group_GSE216841 = rep_by_len(c("MN", "Control"), list(SID_MN, SID_Control))
expM_list = list2(expM_GSE108109, expM_GSE216841)
names(expM_list) = subString(names(expM_list), 2, "_")
names(expM_list)
- 'GSE108109'
- 'GSE216841'
group_merge = Reduce(c, sapply(paste0("group_", names(expM_list)), get))
table(group_merge)
group_merge
Control MN
14 56
set_image(9, 10)
obj = run_ComBat(expM_list, group_merge, out_name = "./test/run_ComBat/01_Combat")
Warning message in MASS::cov.trob(data[, vars]):
"Probable convergence failure"
Warning message in MASS::cov.trob(data[, vars]):
"Probable convergence failure"
Warning message in MASS::cov.trob(data[, vars]):
"Probable convergence failure"
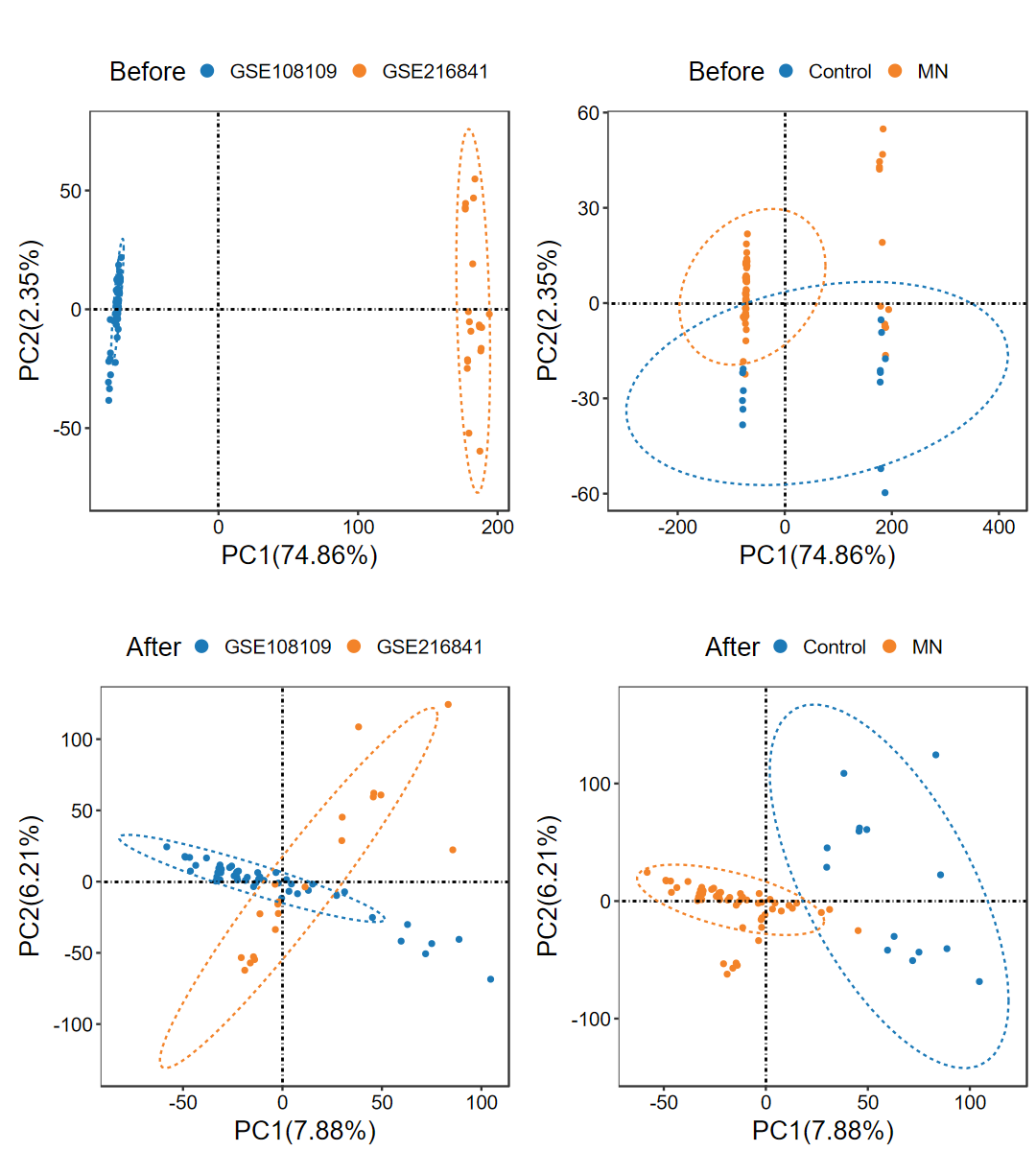
# 提取结果
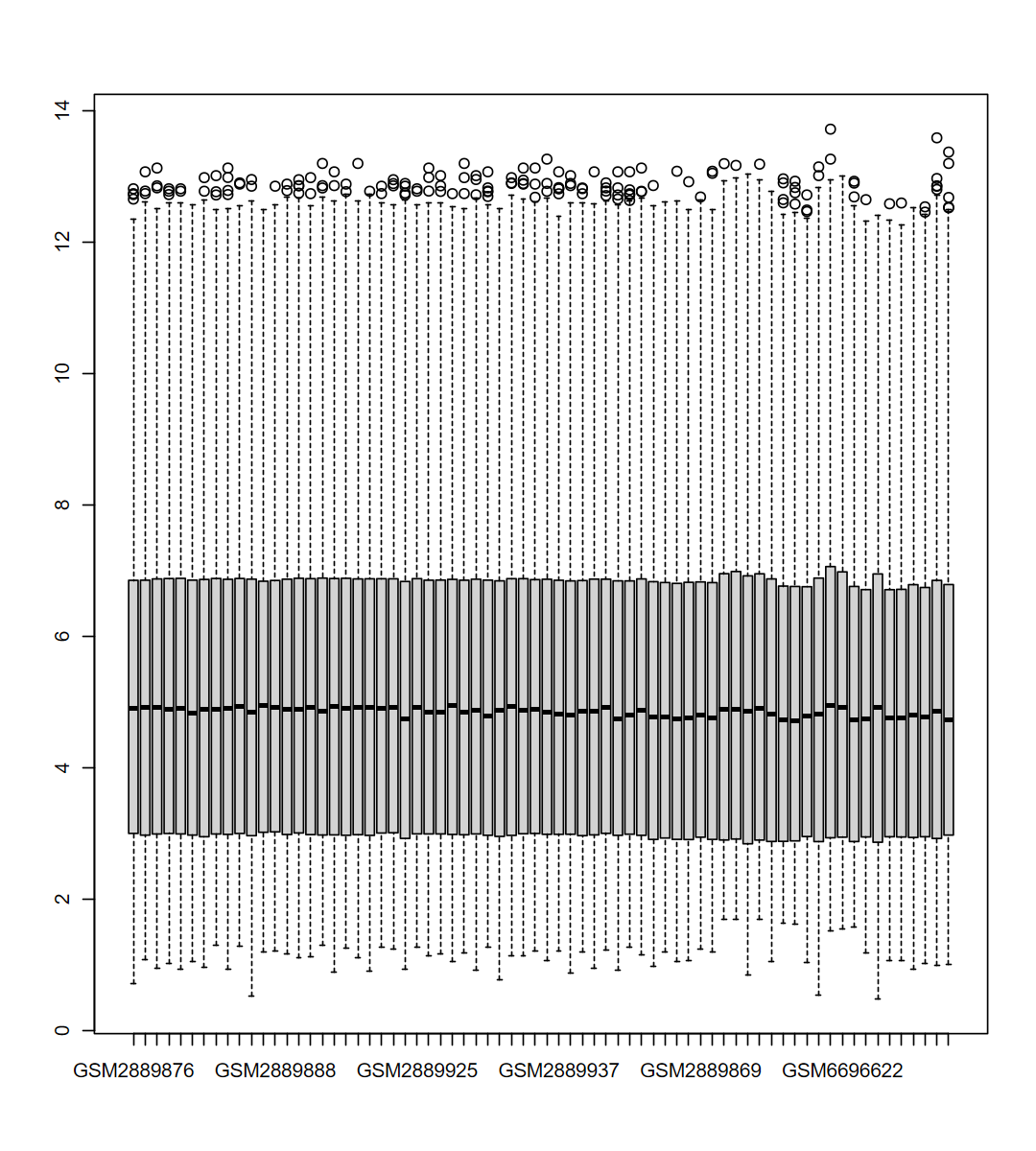
expM = obj$expM
group = obj$group
boxplot(expM)
table(group)
group
Control MN
14 56
search_GEO: 数据集检索
- 这个功能是V1.9最主要和重要的功能, 它可以让数据集检索结果的筛选变得极其高效!
- 结合AI人工智能, 只需简单的三步就能获取GEO数据库中你想要的数据集
第一步: 获取检索词
- 比如你需要检索以下几个关键词的数据: 人类阿尔兹海默症海马体组织的bulk转录组数据
- 首先定义keywords, 然后使用get_GEO_query_promot生成AI提示词, 把提示词粘贴给AI即可获得检索词
- ChatGPT生成的检索词感觉最为靠谱
- 当然如果你自己会写检索词也可以自己写
- 检索词只有一个要求: 不能包含单引号!
keywords = c("阿尔兹海默症", "转录组(RANseq和微阵列)", "海马体组织", "人类")
get_GEO_query_promot(keywords)
我需要检索GEO数据集,请编写符合下面条件的英文单行检索词,同义词尽可能地多,宁愿多检索也不能漏掉,物种要这样写"Homo sapiens"[porgn], 条件: 阿尔兹海默症、转录组(RANseq和微阵列)、海马体组织、人类。 回答仅给出检索词就行, 放在代码块里检索词, 最后一定要仔细检查去除检索词里的单引号
第二步: 下载数据集信息
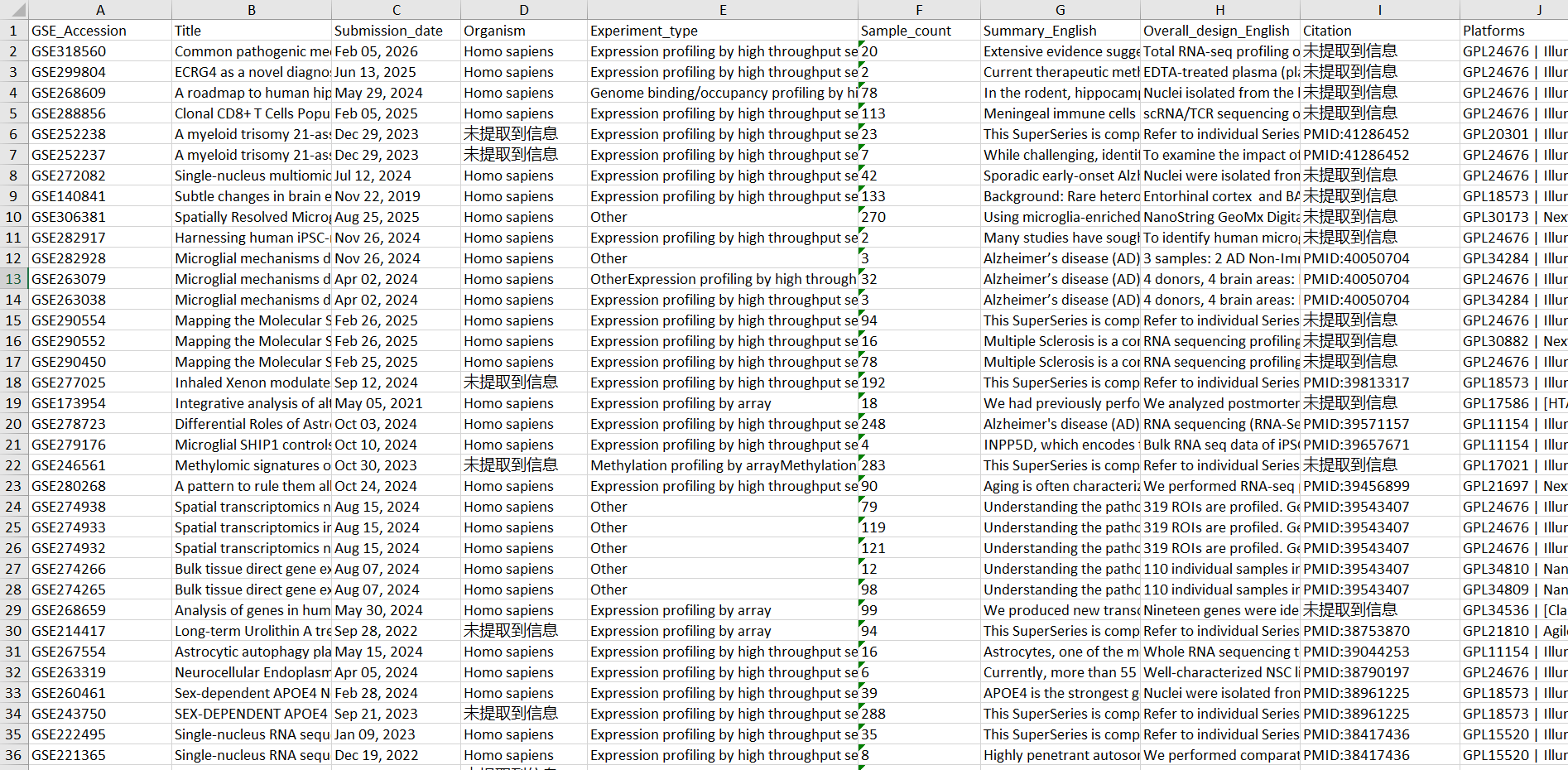
- 之后, 将AI或自己编写的检索词提供给函数search_GEO, 它就可以全自动获取满足检索条件的所有数据集的信息, 基本上是GSE界面的所有信息
- 并且分别保存为txt和xlsx格式, 前者提供给AI解析筛选, 后者我们也可以进行查看和筛选
search_term = '("Alzheimer disease") AND (transcriptome OR transcriptional OR "gene expression" OR "high throughput sequencing" OR microarray) AND (hippocampus) AND "Homo sapiens"[porgn]'
search_GEO(search_term, out_name = "./test/阿尔兹海默_海马体_转录组数据")
>>>>> 步骤1: 构建检索链接,获取符合条件的UID ...
>>>>> 成功检索到81个符合条件的UID
>>>>> 步骤2: 提取每个UID对应的完整GSE Accession
>>>>> 步骤3: 下载每个GSE的HTML文件
>>>>> 步骤4: 提取每个GSE的元数据信息
Warning message in file.create(to[okay]):
"cannot create file './test/阿尔兹海默_海马体_转录组数据.xlsx', reason 'Permission denied'"
>>>>> 检索结果已保存到: ./test/阿尔兹海默_海马体_转录组数据.xlsx
>>>>> 检索结果已保存到: ./test/阿尔兹海默_海马体_转录组数据.txt
第三步: 让AI精确筛选想要的数据集
- 获取每个数据集的信息后, 就可以扔给AI让它帮我们筛选数据了, 几十分钟甚至几个小时的工作AI十几秒就可以完成了
- 首先贴心地为大家准备了生成提示词地函数: get_GEO_summary_promot, 大家也可以跟AI随便说你想要的数据
- 然后将上面生成的txt文件和提示词扔给AI, 它就很快的帮我们把需要的数据和介绍整理成表格了
- 这里我用的AI是豆包, 因为豆包对文件的处理速度最快, 然最后需要我们手动一一校验是否满足我们的需求
get_GEO_summary_promot(keywords)
从提供的文件中筛选整理严格符合下面条件的数据集成表格, 首先总结满足条件的数据集, 然后给出表格包含下面几列:GSE编号、核心研究内容、关键技术、样本信息: 条件: 阿尔兹海默症、转录组(RANseq和微阵列)、海马体组织、人类
-
txt
-
-
xlsx
-
-
AI筛选的结果
-

-
做之前根本不敢想, 这竟然真的能够实现, 谁懂这个R语言+AI的含金量!
安装
# 当前目录的文件
cbind(dir())
| 00_GEO_data |
| 01_安装.R |
| 02_使用.R |
| AnnoProbe.zip |
| annot_dir |
| fastGEO安装和使用教程.html |
| fastGEO安装和使用教程.ipynb |
| fastGEO安装和使用教程.md |
| fastGEO安装和使用教程.R |
| fastGEO安装和使用教程.Rmd |
| fastGEO安装和使用教程.Rproj |
| fastR_1.5.0.tar.gz |
| fastR_1.8.0.tar.gz |
| Heatmap_DEG.pdf |
| Heatmap_DEG.png |
| test |
| test.R |
| test.txt |
| tmp_GSE_html |
| 阿尔兹海默_海马体_转录组数据.txt |
| 阿尔兹海默_海马体_转录组数据.xlsx |
| 测试案例 |
- 双击html文档可以打开网页版的此教程, 可以浏览, 但不能运行
- 安装和使用视频教程: R包fastGEO-GEO数据库快速下载探针注释
- 双击项目文件
fastGEO安装和使用教程.Rproj, 打开项目 - 右下角Files面板点击R脚本, 运行安装代码
必须安装的
- 执行基础的功能所必须的
# if (!require("BiocManager", quietly = TRUE))
# install.packages("BiocManager")
# BiocManager::install("GEOquery")
# BiocManager::install("limma")
# devtools::install_github("jmzeng1314/AnnoProbe")
# install.packages("data.table", repos="https://rdatatable.gitlab.io/data.table")
# devtools::install_github("hadley/readr")
# install.packages("stringr")
# install.packages("httr")
# install.packages("rvest")
# install.packages("shiny")
# install.packages("DT")
建议安装的
- 这些可能不太容易安装, 尽量安装一下
# BiocManager::install("org.Hs.eg.db")
# BiocManager::install("org.Mm.eg.db")
# BiocManager::install("clusterProfiler")
安装我的工具包 fastR
- 有很多函数是依赖我的这个工具包的
- 当前文件夹没有安装包的需要付费获取安装包:
- 直接添加微信转账50, 发送fastR和fastGEO的压缩包
- 以前是20, 老用户依然可免费获取
- 很多资料后面都会慢慢涨价, 早买会便宜很多!
- 购买后可进入交流群,后续版本更新和问题答疑都在群里进行

# 自动寻找当前目录下的压缩包
file_fastR = grep("^fastR.*.gz", dir(), value = TRUE)
file_fastR
- 'fastR_1.5.0.tar.gz'
- 'fastR_1.8.0.tar.gz'
# 如果你正在使用要安装或更新的R包, 需要先取消加载, 取消占用
# detach("package:fastGEO", unload = TRUE)
# 强制进行安装更新
# install.packages(file_fastR, repos = NULL, upgrade = FALSE, force = TRUE)
安装 fastGEO
# ## 如果以前安装过, 可能本地存在注释文件, 如果有则需要备份
# ## 重新安装前保存预先存在的注释文件
# anno_file = paste0(.libPaths(), "/fastGEO/data/anno_obj.RData")
# anno_file_temp = paste0("temp-", basename(anno_file))
# anno_file_exists = FALSE
# if(file.exists(anno_file)){
# file.copy(anno_file, anno_file_temp)
# anno_file_exists = TRUE
# }
# anno_file_exists
# ## 安装fastGEO
# file_fastGEO = grep("^fastGEO.*.gz", dir(), value = TRUE)
# install.packages(file_fastGEO, repos = NULL, upgrade = FALSE, force = TRUE)
# ## 重新拷贝注释文件
# if(anno_file_exists){
# file.copy(anno_file_temp, anno_file)
# unlink(anno_file_temp)
# }
- 安装完之后按照上一节的内容自行测试就行了
更新版本的安装
- 对于小版本的细微改动, 我将只提供更新后的R包压缩包, 大家可以将压缩包放到教程目录下, 重新运行安装代码即可
- 或者直接指定安装包路径进行安装
- 其中
file_pkgs是R包压缩包的绝对路径
# install.packages(file_pkgs, repos = NULL, upgrade = FALSE, force = TRUE)

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐


 18
18 0
0- 0



 已为社区贡献3条内容
已为社区贡献3条内容

所有评论(0)