R包fastEnrich开发2: 一键运行和可视化GO/KEGG/DO/Reactome富集分析及美化图片
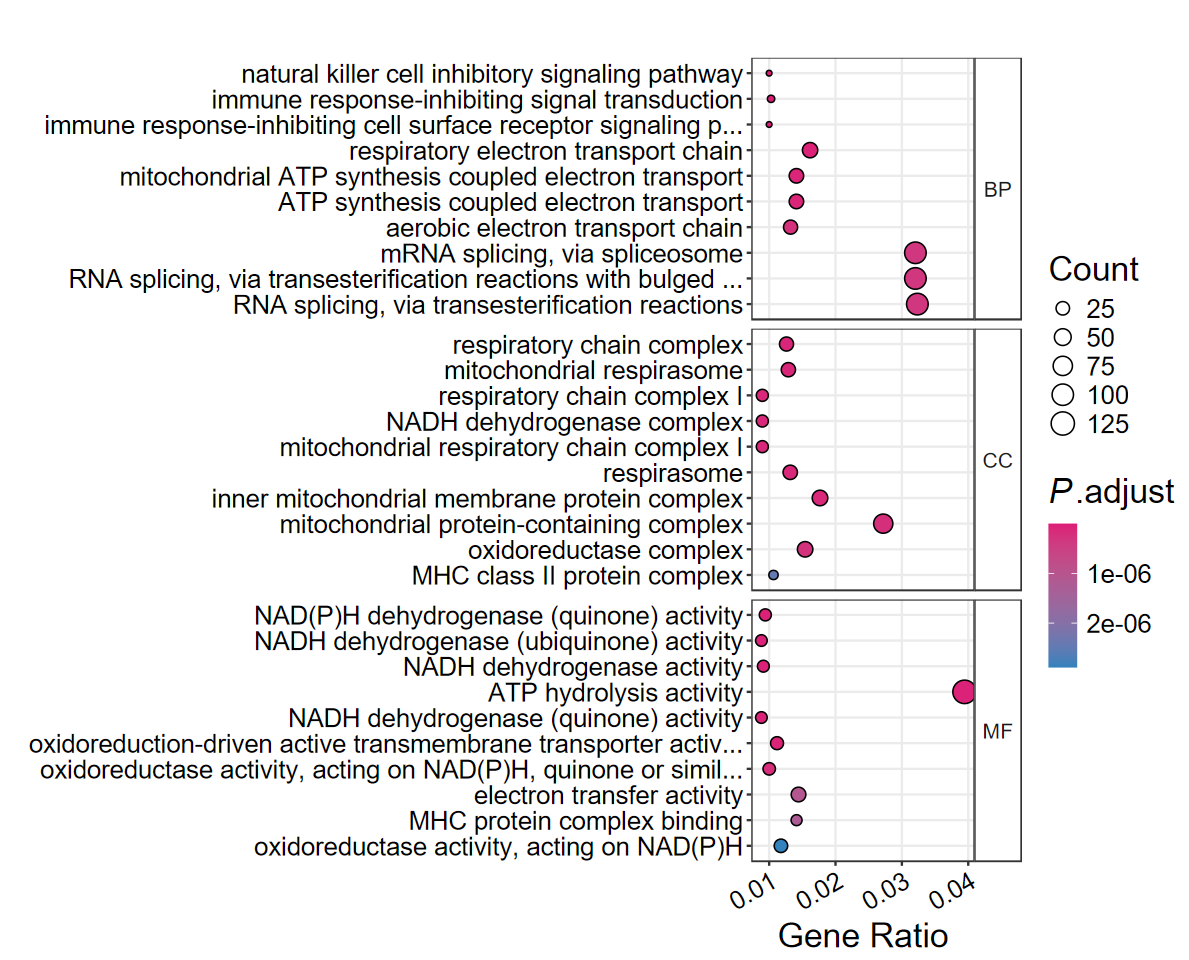
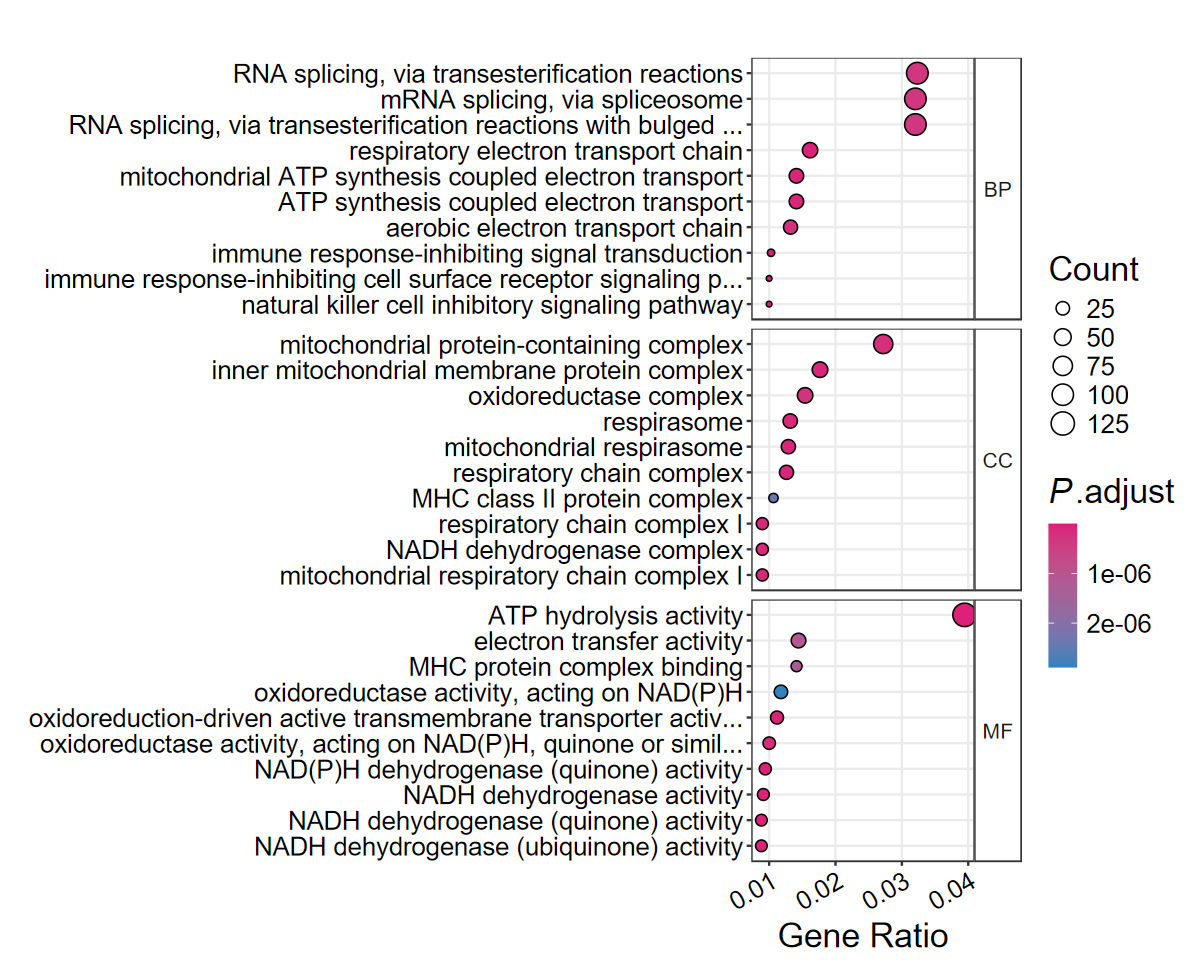
CC共富集到93个通路,富集的top5通路分别为:呼吸链复合体(respiratory chain complex)、线粒体呼吸体(mitochondrial respirasome)、线粒体呼吸链复合体I(mitochondrial respiratory chain complex I)、NADH脱氢酶复合体(NADH dehydrogenase complex)和呼吸链复合体I(respir
开发背景
- 生信分析中, 富集分析是非常常见的分析之一, 而且种类繁多, 至少包括各种通路富集分析、GSEA、单基因GSEA、GSVA、ssGSEA
- 而且基因ID的准备也需要转来转去, 很是麻烦
- 其次可视化的方式也非常多, 美观度参差不齐
- 最后对与富集结果的报告, 每次要复制粘贴翻译编辑很少麻烦
- 因此, 很久之前就准备开发个R包, 专门快速实现和可视化所有的富集分析相关的分析
- 后续会慢慢开发, 这次第二次发布 【目前定价是100】, 第一次发布费用是50, 后续至少会涨到200
- 所以需要的就赶紧买, 功能会非常多, 绝对划算, 微信 【直接转账】 给我即可
- 买过我的R包fastGEO V1.8.0 快速下游分析,多数据集批量下载处理,火山图优化都知道, R包的价格会随之更新的内容增加而上调了, 由20调到50再到100。
- 所以, 后续的R包也是一样, 越早买越便宜, 可永久免费获取最新版本!并且有交流群
# # 载入富集分析函数, 作者测试用的
# library(fastR)
# fun_dir = "W:/02_Study/R_build/build/05_fastEnrich/functions"
# invisible(sapply(lf("rf", fun_dir), source))
Loading required package: ggplot2
Warning message:
"package 'ggplot2' was built under R version 4.4.3"
# 加载R包
library(fastEnrich)
功能演示
run_GO: 快速GO富集
-
首先解决的就是ID自动转换问题
-
我们常用的是SYMBOL, 而GO要使用ENSEMBL, KEGG要使用ENTREZID 。。。
-
run_GO和run_KEGG可以根据输入基因的特征自动判断并转换ID, 无需再手动转换
-
需要注意的是目前只支持人和小鼠的自动富集分析, 因为我不做其他的物种
-
之后就是使用clusterProfiler进行富集分析了
-
然后解决的就是输出表格geneID这一列了, 原始表格依然是ENSEMBL或者ENTREZID, 然而我们还是需要SYMBOL进行快速查看, 所以就再给它转回去。。。
-
最后就是富集结果报告的自动生成, 报告富集到了多少通路, BP CC MF分别有多少,top5是哪些通路,并且进行翻译和文本拼接
-
这里使用limma的差异表达结果的差异基因进行测试
# 进入输出目录
setwd("test")
DEG_tb = read.csv("DEG_tb.csv", row.names = 1)
DEGs = rownames(DEG_tb)[DEG_tb$DEG != "Not_Change"]
hd(DEGs)
Type: character Dim: 3702
[1] "MMP1" "SPP1" "BPIFB1" "CP" "ND6"
- 物种默认为人类(human), 可通过参数
OrgDb指定物种或注释R包 - 目前支持的物种有: human(默认)、mouse(小鼠)和rat(大鼠),可以直接指定物种名字就行了
- 而其他物种就需要指定对应的R包了, 比如斑马鱼
org.Dr.eg.db
ego = run_GO(DEGs, out_name = "GO_enrich")
>>>>> 校验并载入基因组注释R包: org.Hs.eg.db
>>>>> 自动转换基因ID为ENSEMBL
>>>>> 运行GO富集分析, ONTOLOGY: ALL
>>>>> 生成富集报告
>>>>> 运行结束, 共显著富集到727个通路, BP:559 CC:93 MF:75
hd(ego, 5, 10)
Type: enrichResult Dim: 727 × 10
ONTOLOGY ID Description GeneRatio BgRatio pvalue p.adjust
GO:0002769 BP GO:0002769 natural killer cell inhibitory signaling pathway 34/3402 36/21288 3.281964e-25 2.102098e-21
GO:0002765 BP GO:0002765 immune response-inhibiting signal transduction 35/3402 63/21288 6.761106e-13 2.165244e-09
GO:0002767 BP GO:0002767 immune response-inhibiting cell surface receptor signaling pathway 34/3402 61/21288 1.268932e-12 2.709170e-09
GO:0022904 BP GO:0022904 respiratory electron transport chain 55/3402 140/21288 2.577823e-11 4.127739e-08
GO:0042773 BP GO:0042773 ATP synthesis coupled electron transport 48/3402 117/21288 7.858191e-11 8.388619e-08
qvalue
GO:0002769 1.719058e-21
GO:0002765 1.770698e-09
GO:0002767 2.215511e-09
GO:0022904 3.375592e-08
GO:0042773 6.860063e-08
geneID
GO:0002769 KIR2DL1
GO:0002765 CD33/KIR2DL1
GO:0002767 KIR2DL1
GO:0022904 ND6/DLD/NDUFA5/CCNB1/VPS54/NDUFS1/ETFDH/SLC25A12/PUM2/GPD2/SDHD/NDUFA7/NDUFS4/NDUFB4/COX4I2/COX7A2/COX4I1/NDUFB9/NDUFA12/GPD1/NDUFS5/NDUFB10/SDHB/NDUFA3/COX7A1/NDUFC2/DGUOK/NDUFA6/UQCR10/SDHC/NDUFS7/COX6B1/NDUFC2-KCTD14/DNAJC15/NDUFA2/COX6A1/COX8A/COX5B
GO:0042773 ND6/DLD/NDUFA5/CCNB1/NDUFS1/SDHD/NDUFA7/NDUFS4/NDUFB4/COX4I2/COX7A2/COX4I1/NDUFB9/NDUFA12/NDUFS5/NDUFB10/SDHB/NDUFA3/COX7A1/NDUFC2/DGUOK/NDUFA6/UQCR10/SDHC/NDUFS7/COX6B1/NDUFC2-KCTD14/DNAJC15/NDUFA2/COX6A1/COX8A/COX5B
Count
GO:0002769 1
GO:0002765 2
GO:0002767 1
GO:0022904 38
GO:0042773 32
- 经过V2版本的更新, 所有富集分析的返回的对象保持原有类型, 即enrichResult, 这样就能适配其他R包下游的分析函数了
- 我觉得通路名换行会好丑, 宁愿直接省略…
- 因此我下面也会进行改造
class(ego)
‘enrichResult’
set_image(10, 7)
barplot(ego, showCategory = 20, label_format = 100)
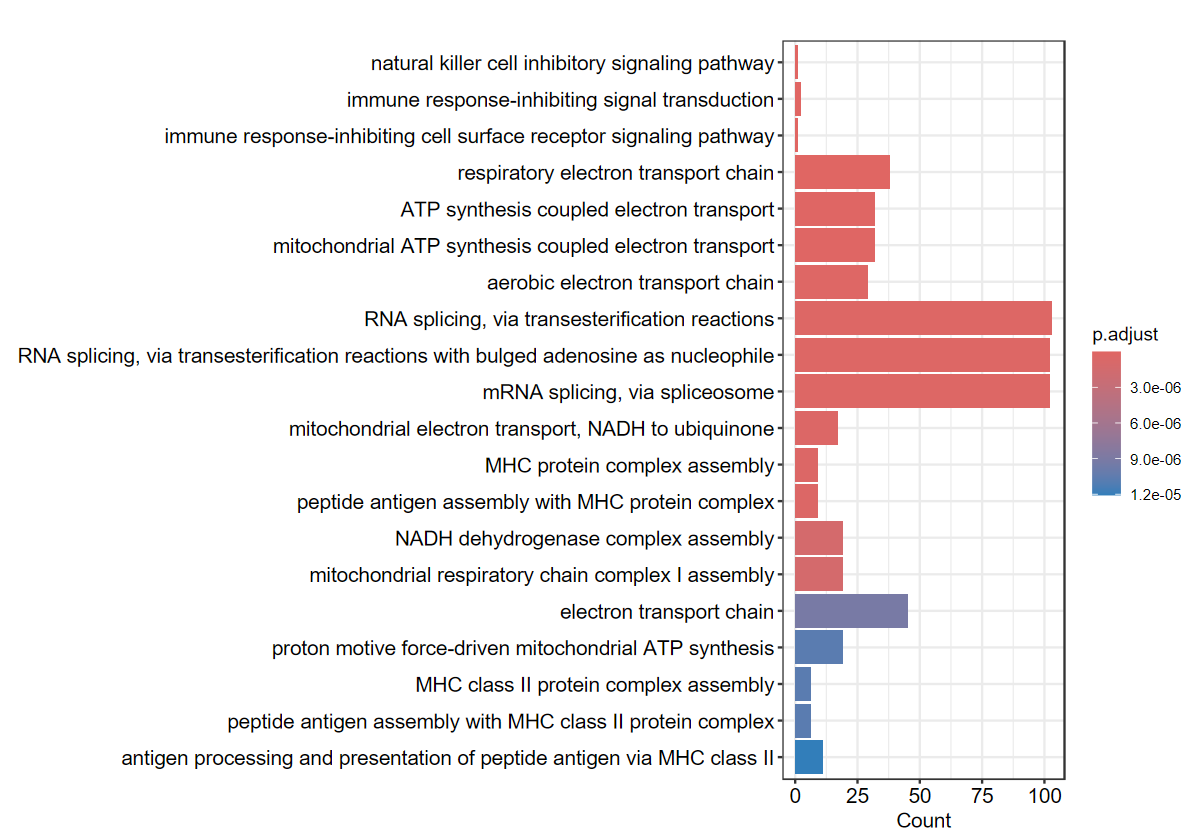
dotplot(ego, showCategory = 20, label_format = 100)
cnetplot(ego, categorySize = "pvalue")

run_KEGG: 快速KEGG富集
- 首先解决的还是是ID自动转换问题, KEGG要使用ENTREZID
- 我们常用的是SYMBOL, 而GO要使用ENSEMBL 。。。
ekegg = run_KEGG(DEGs, out_name = "KEGG_enrich")
>>>>> 校验并载入基因组注释R包: org.Hs.eg.db
>>>>> 自动转换基因ID为ENTREZID
>>>>> 运行KEGG富集分析
>>>>> 生成富集报告
>>>>> 共富集到57个通路!
run_DO: 快速DO富集
edo = run_DO(DEGs, out_name = "DO_enrich")
>>>>> 校验并载入基因组注释R包: org.Hs.eg.db
>>>>> 自动转换基因ID为ENTREZID
>>>>> 运行DO富集分析
>>>>> 生成富集报告
>>>>> 运行结束, 共显著富集到100个通路
run_Reactome: 快速Reactome富集
eReactome = run_Reactome(DEGs, out_name = "Reactome_enrich")
>>>>> 校验并载入基因组注释R包: org.Hs.eg.db
>>>>> 自动转换基因ID为ENTREZID
>>>>> 运行Reactome富集分析
>>>>> 生成富集报告
>>>>> 运行结束, 共显著富集到41个Reactome通路
report_pathway - 自动化报告
- run_GO会自动调用report_pathway_GO, 而后者调用的是report_pathway
# 查看报告
readLines("GO_enrich_report.txt")
‘GO富集分析表明,共富集到727个通路。其中BP共富集到559个通路,富集的top5通路分别为:自然杀伤细胞抑制信号通路(natural killer cell inhibitory signaling pathway)、免疫反应抑制信号转导(immune response-inhibiting signal transduction)、免疫反应抑制剂细胞表面受体信号通路(immune response-inhibiting cell surface receptor signaling pathway)、呼吸电子传递链(respiratory electron transport chain)和ATP合成偶联电子传递(ATP synthesis coupled electron transport)。MF共富集到75个通路,富集的top5通路分别为:NAD(P)H脱氢酶(醌)活性(NAD§H dehydrogenase (quinone) activity)、NADH脱氢酶活性(NADH dehydrogenase activity)、NADH-脱氢酶(泛醌)活性(NADH dehydrogenase (ubiquinone) activity)、ATP水解活性(ATP hydrolysis activity)和NADH-脱氢酶(醌(NADH dehydrogenase (quinone) activity)。CC共富集到93个通路,富集的top5通路分别为:呼吸链复合体(respiratory chain complex)、线粒体呼吸体(mitochondrial respirasome)、线粒体呼吸链复合体I(mitochondrial respiratory chain complex I)、NADH脱氢酶复合体(NADH dehydrogenase complex)和呼吸链复合体I(respiratory chain complex I)。’
- 直接粘贴进分析报告就行了, 手动整理就问你得多久,逆天功能!
- 当然翻译是务必需要人工矫正的。。。
dotplot_GO: GO富集分析气泡图
-
默认每个ONTOLOGY绘制10个, 也可以选择绘制所有的, 当然这种情况是你已经对ego做了筛选, 不然一般会很多
-
自动化输出图片有个问题是图片宽度的设置, 这里也自动根据字符长度最长的通路进行自动调整宽度了, 当然如果我自动调整的不合适你可以再指定参数
w -
设置
out_name指定输出文件名, 不带后缀 -
由于有的通路名实在太长, 放在文章里实在不合适, 但又不想删掉, 解决策略就是省略后面一部分, 所以自动设置了最长显示字符宽度为60, 可以通过
max_omit调整和omit = FALSE进行关闭自动省略字符功能。 -
默认参数
set_image(10, 8) # set_image仅控制在jupyter里图片的宽度, 不影响Rstudio的显示和文件的输出
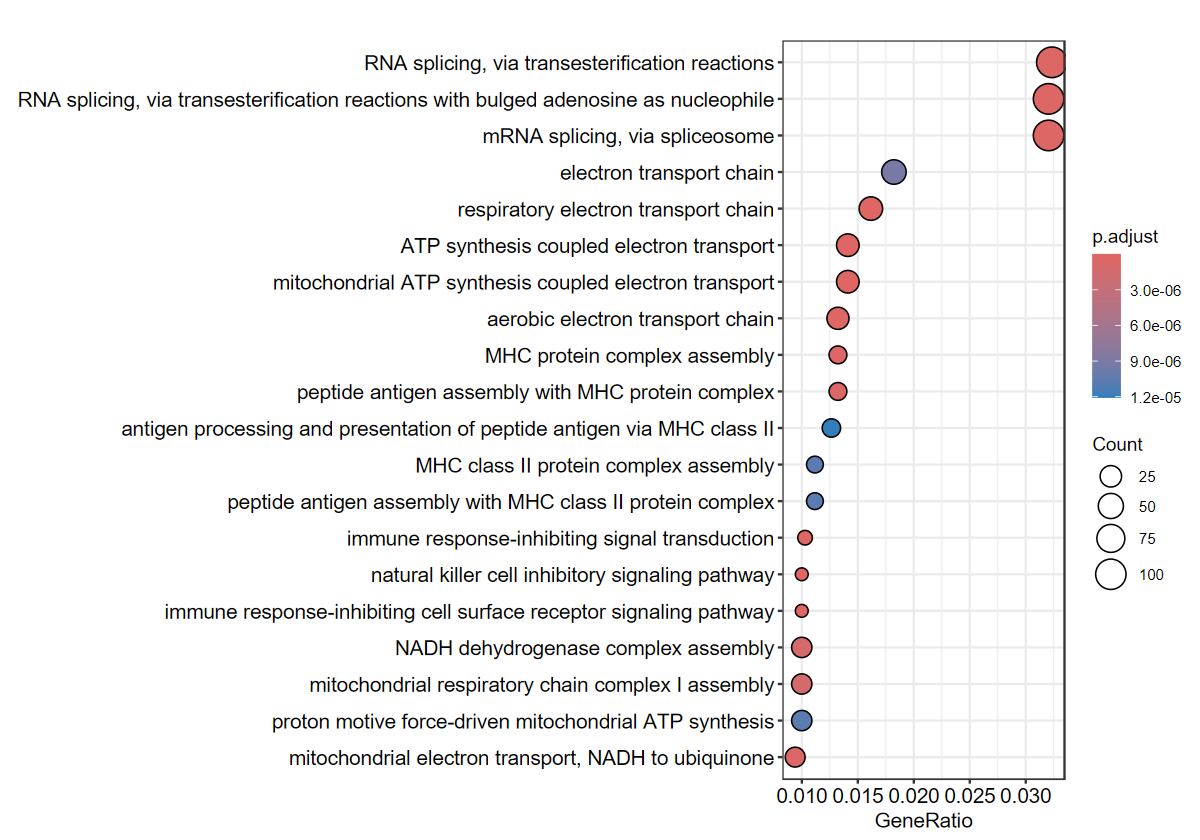
dotplot_GO(ego, out_name = "GO_dotplot")
- 思来想去, 虽然气泡按大小从左到右排序会更好看, 但是我们更关注的以及要描述的还是P值比较小的
- 所以这种情况下如果按Ratio排序会造成图文不一致的问题, 你说某个通路最显著, 一看图不是在最上面, 还要找呀找。。。
- 因此, 我还是将P值作为排序条件, P值越小排在最上面
- 如果你想要按ratio排序, 只需要添加
sort = "ratio"就行了
dotplot_GO(ego, out_name = "GO_dotplot2", sort = "ratio")
- 如果想通路少一些, 调整
ntop
set_image(10, 8) # set_image仅控制在jupyter里图片的宽度, 不影响Rstudio的显示和文件的输出
dotplot_GO(ego, out_name = "02_GO_dotplot3", ntop = 5)
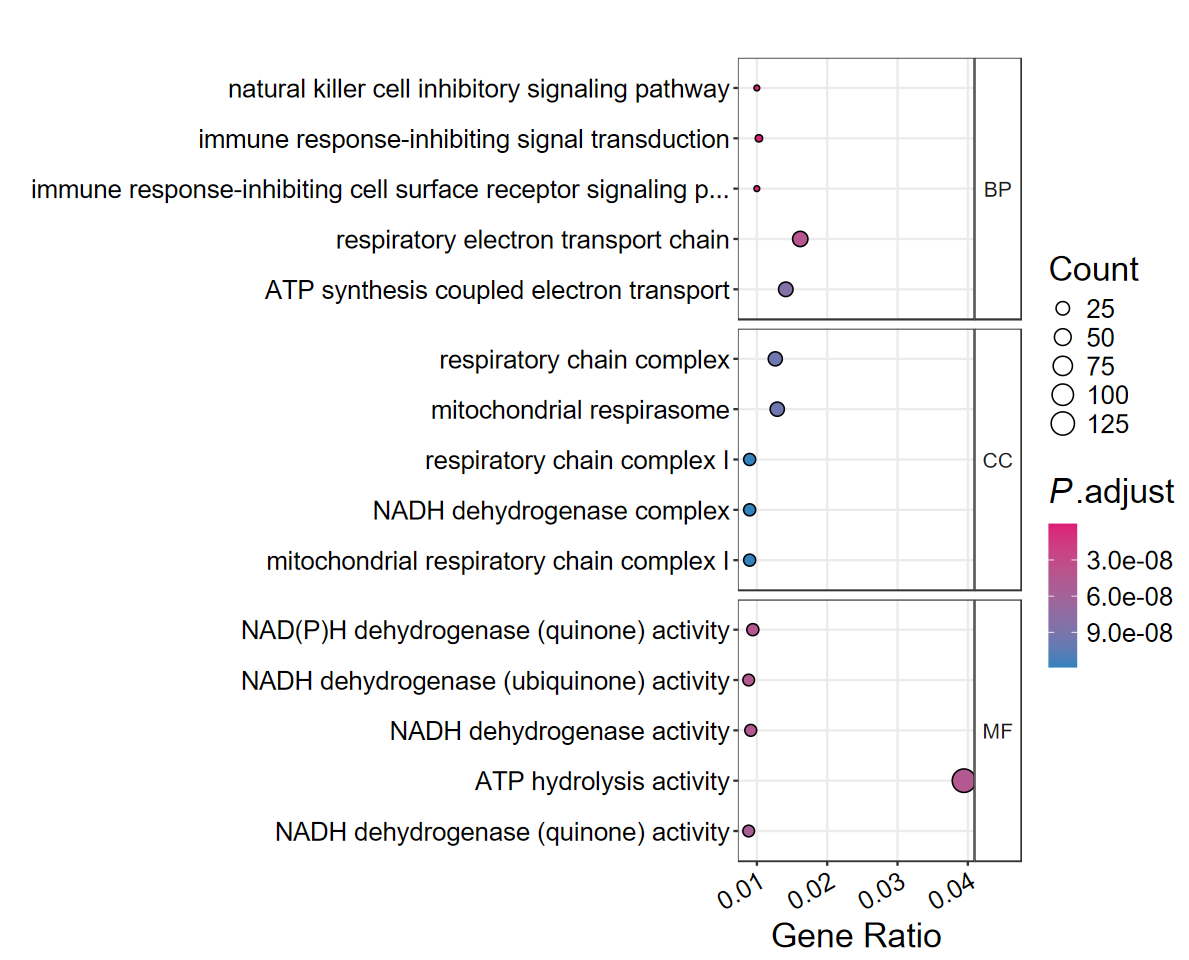
- 不进行字符省略
set_image(13, 8)
dotplot_GO(ego, out_name = "GO_dotplot4", omit = FALSE)
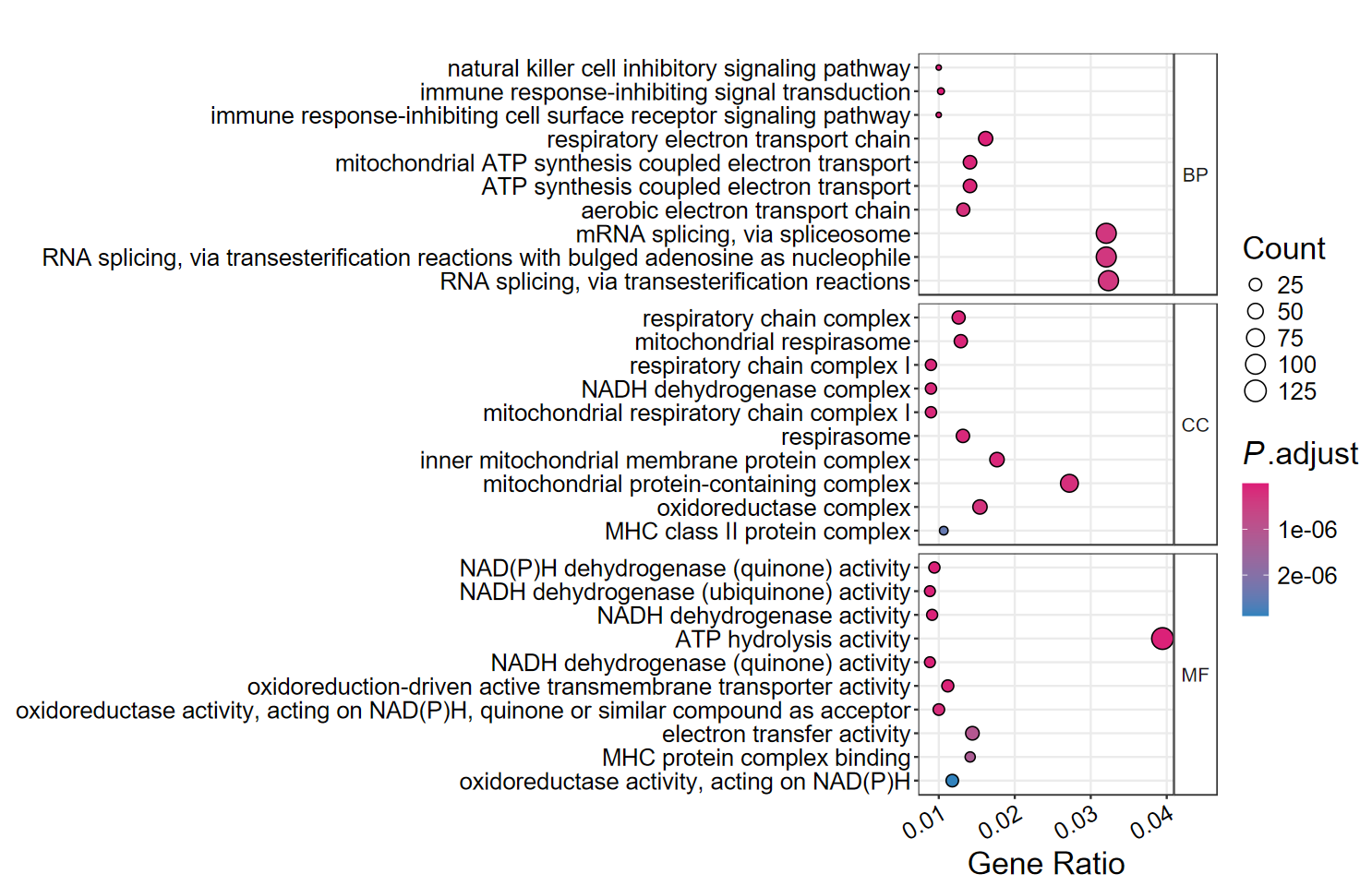
- 如果矫正的通路太少了, 你可以指定
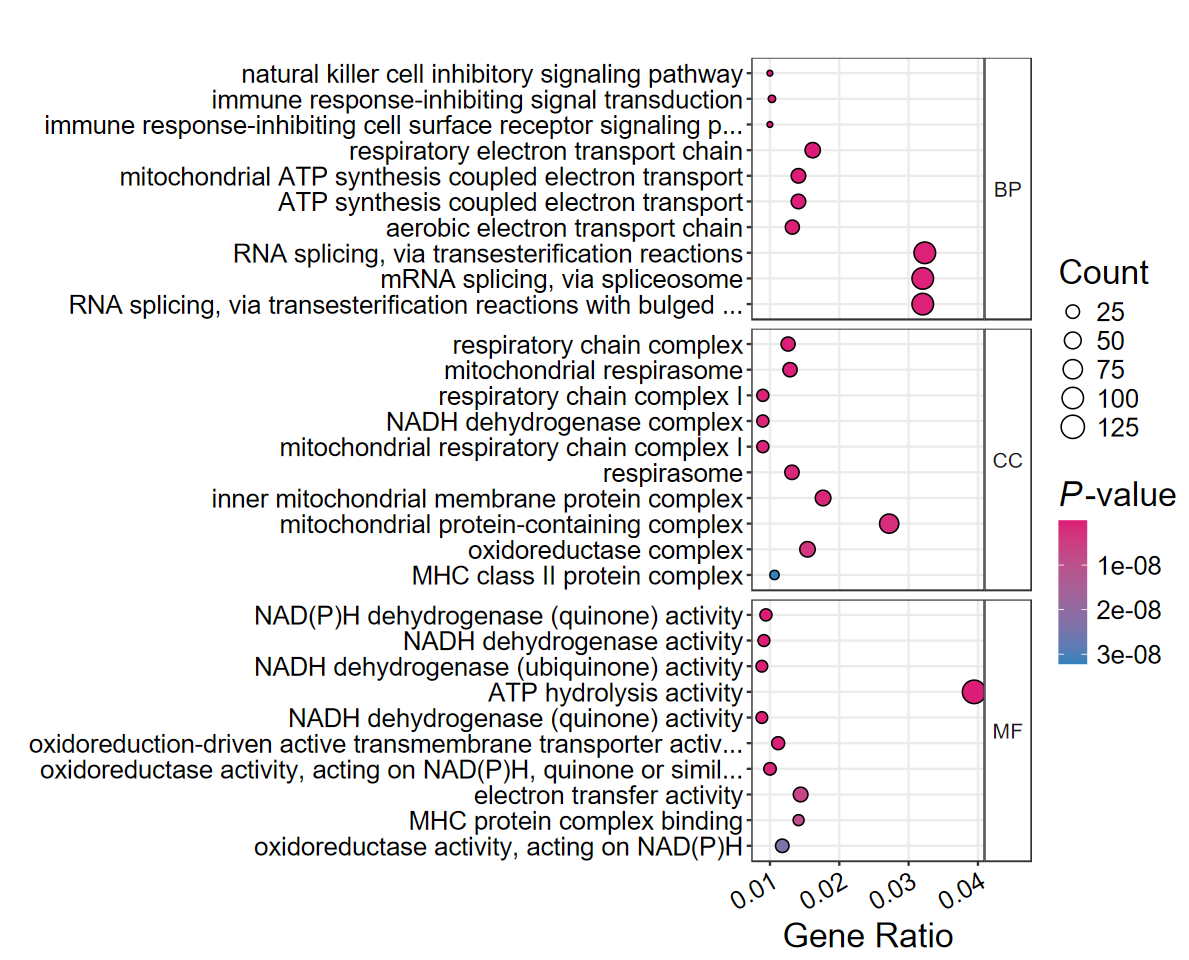
padj = FALSE使用矫正前的显著通路绘图 - 我开发的所有R包相关函数都会支持这个参数
dotplot_GO(ego, out_name = "GO_dotplot", padj = FALSE)
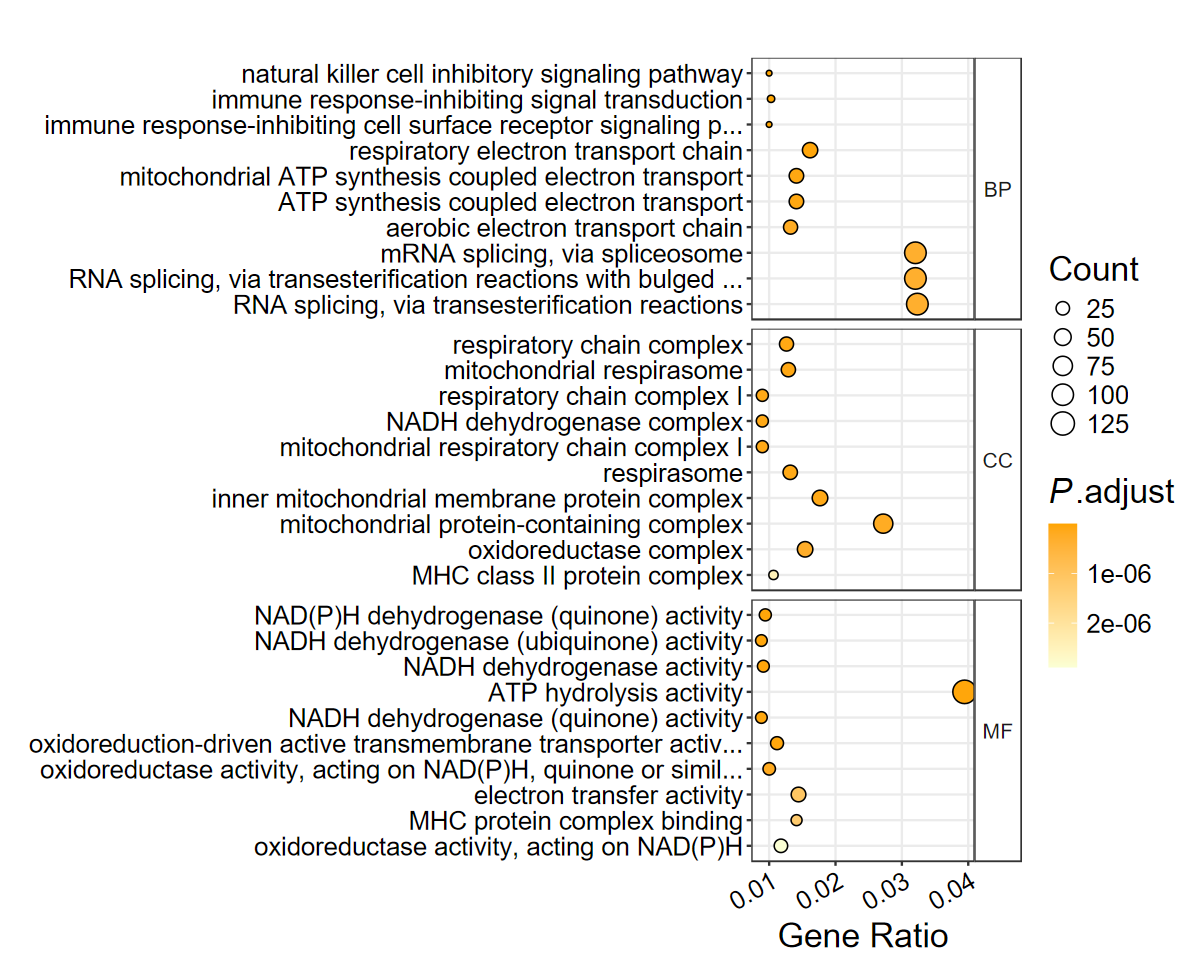
- 自定义气泡颜色, 颜色支持英文和十六进行
set_image(10, 8)
dotplot_GO(ego, out_name = "GO_dotplot5", color_high = "orange", color_low = "#FBFFD4")
- 高亮感兴趣的通路, 并自定义颜色
highlight_paths = c(
"immune response-inhibiting signal transduction",
"MHC class II protein complex",
"NADH dehydrogenase complex",
"MHC protein complex binding",
"mRNA splicing, via spliceosome"
)
dotplot_GO(
ego,
out_name = "GO_dotplot5",
color_high = "orange",
color_low = "#FBFFD4",
highlight = highlight_paths,
highlight_color = "#FFA700"
)
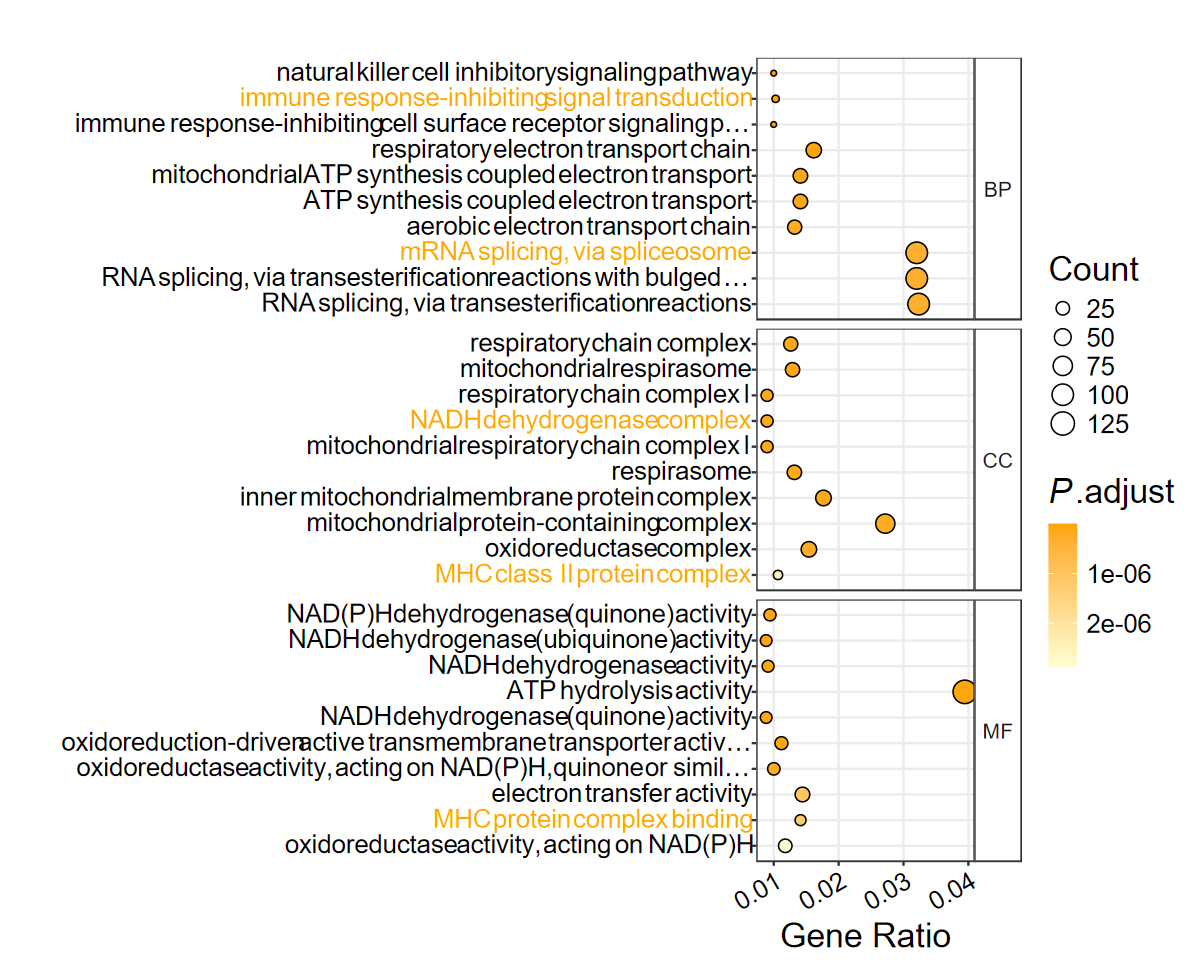
- 还可以添加title, 不过这些基础的ggplot操作都可以后续自由发挥
- 如果有多个气泡图同时展示时, 比如不同的细胞类型、亚群、比较组别, 分别指定不同的颜色排列在一起会好看很多
dotplot_GO(ego,
out_name = "GO_dotplot6",
color_high = "orange",
color_low = "#FBFFD4",
highlight = highlight_paths,
highlight_color = "orange",
title = "A vs B"
)
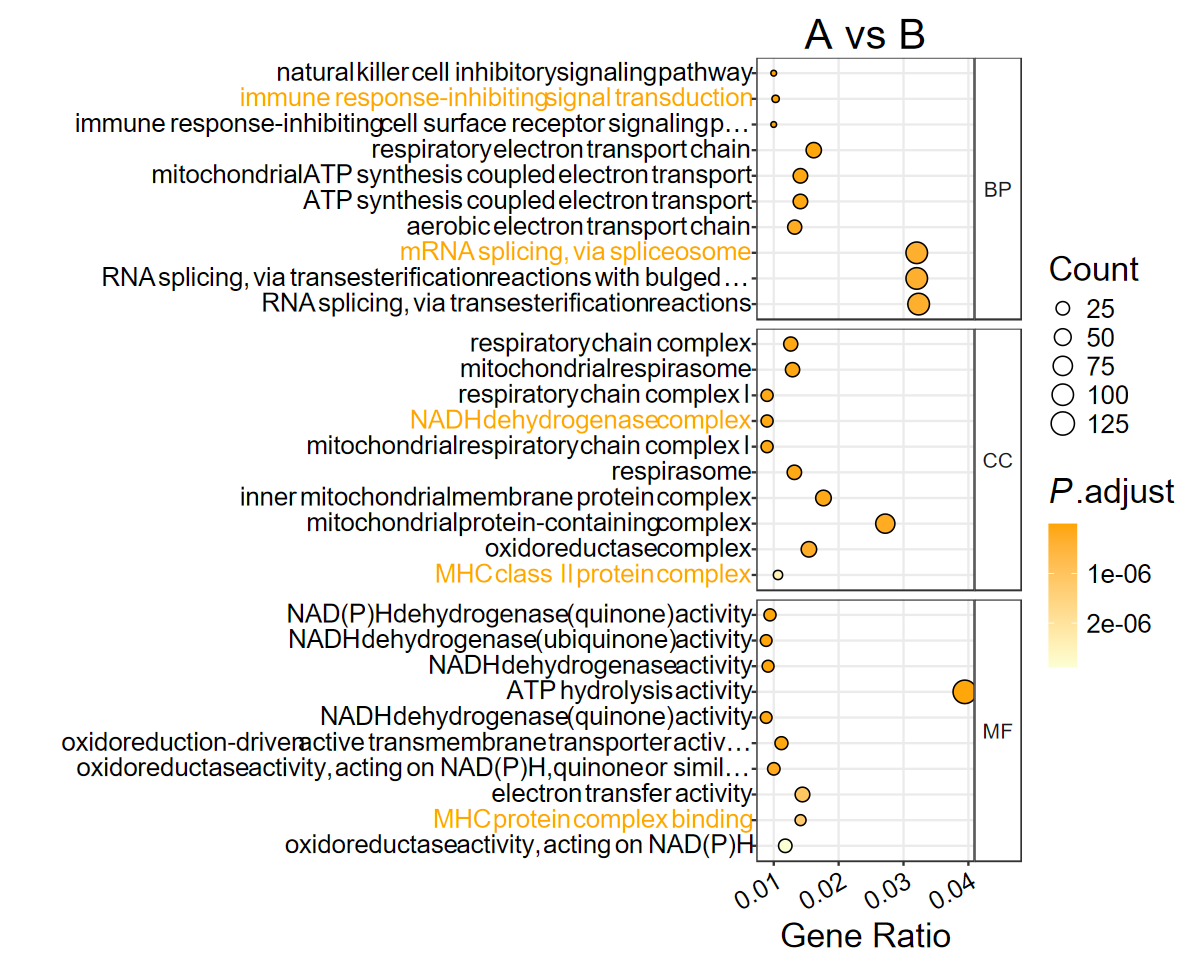
- 也可以手动挑选想要展示的通路, 这时候就不是展示前10了,而是给哪些展示哪些, 设置
show = "all"
# 三种ONTOLOGY分别取 15 10 5进行展示
neach = c(15, 10, 5)
ego2 = Reduce(rbind, lapply(1:length(neach), function(i) split(ego@result, f = ego$ONTOLOGY)[[i]][1:neach[i], ]))
hd(ego2)
Type: data.frame Dim: 30 × 10
ONTOLOGY ID Description GeneRatio BgRatio
GO:0002769 BP GO:0002769 natural killer cell inhibitory signaling pathway 34/3402 36/21288
GO:0002765 BP GO:0002765 immune response-inhibiting signal transduction 35/3402 63/21288
GO:0002767 BP GO:0002767 immune response-inhibiting cell surface receptor signaling pathway 34/3402 61/21288
GO:0022904 BP GO:0022904 respiratory electron transport chain 55/3402 140/21288
GO:0042773 BP GO:0042773 ATP synthesis coupled electron transport 48/3402 117/21288
dotplot_GO(
ego2,
out_name = "GO_dotplot7",
color_high = "orange",
color_low = "#FBFFD4",
highlight = highlight_paths,
highlight_color = "orange",
show = "all",
title = "A vs B"
)
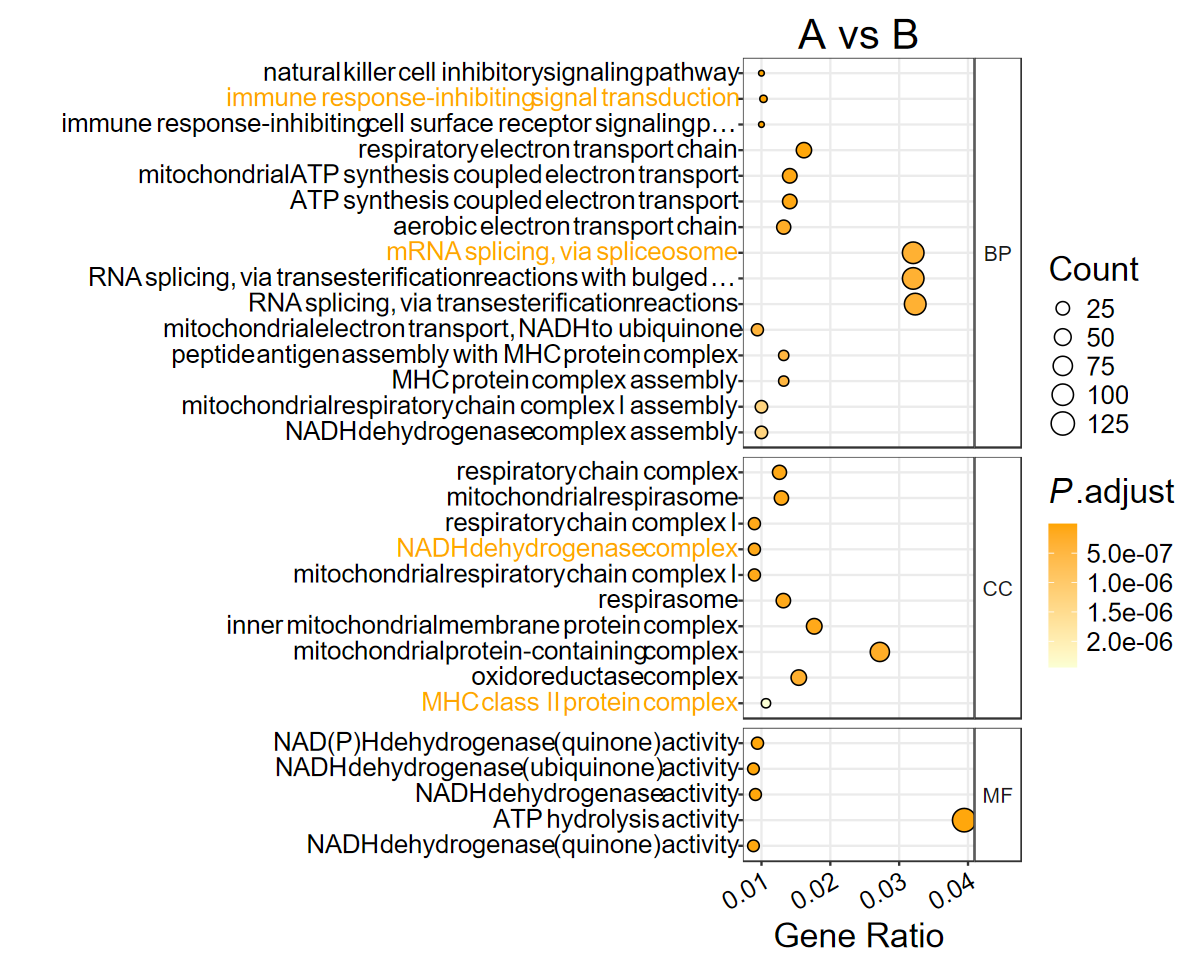
dotplot2: 通用气泡图
dotplot2(ekegg, out_name = "KEGG_dotplot")
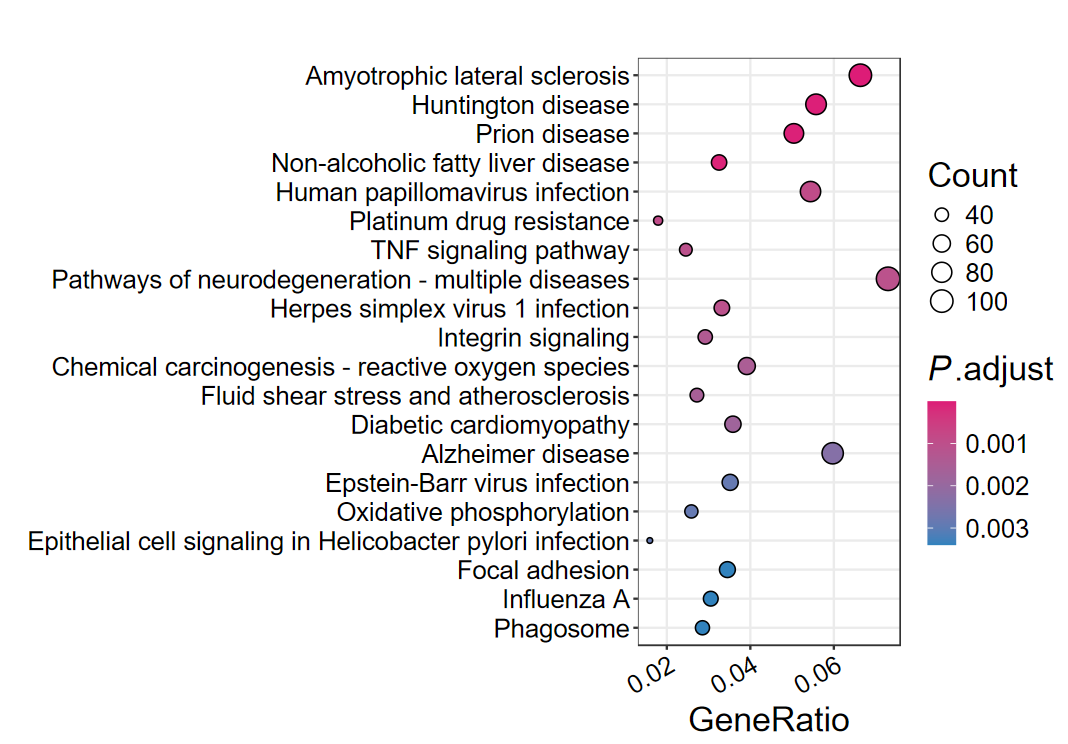
- 自动调整图片的宽高
dotplot2(ekegg, out_name = "KEGG_dotplot_n30", n = 30)
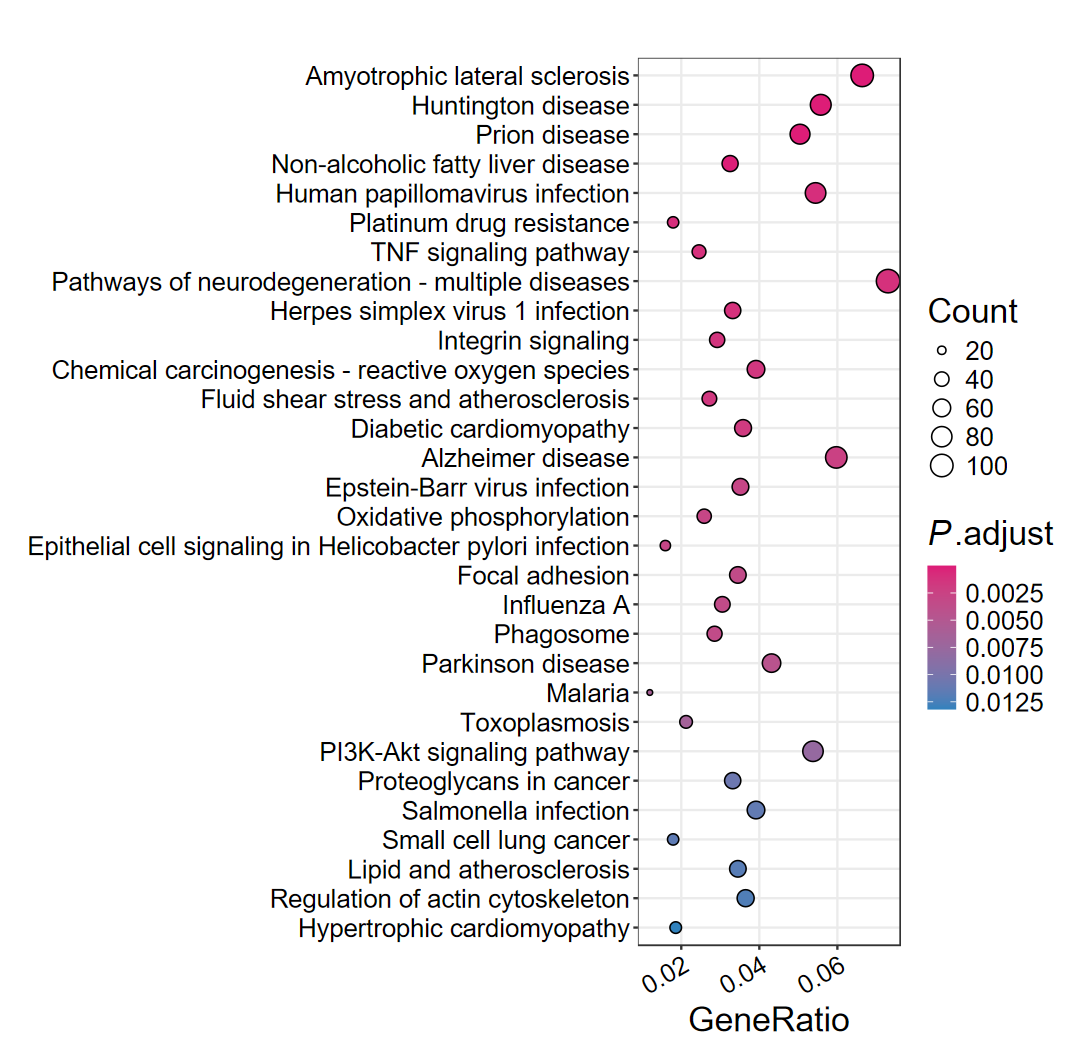
- GO/DO/Reactome结果也是支持的, 所以说是通用的
dotplot2(ego, out_name = "GO_dotplot_base")
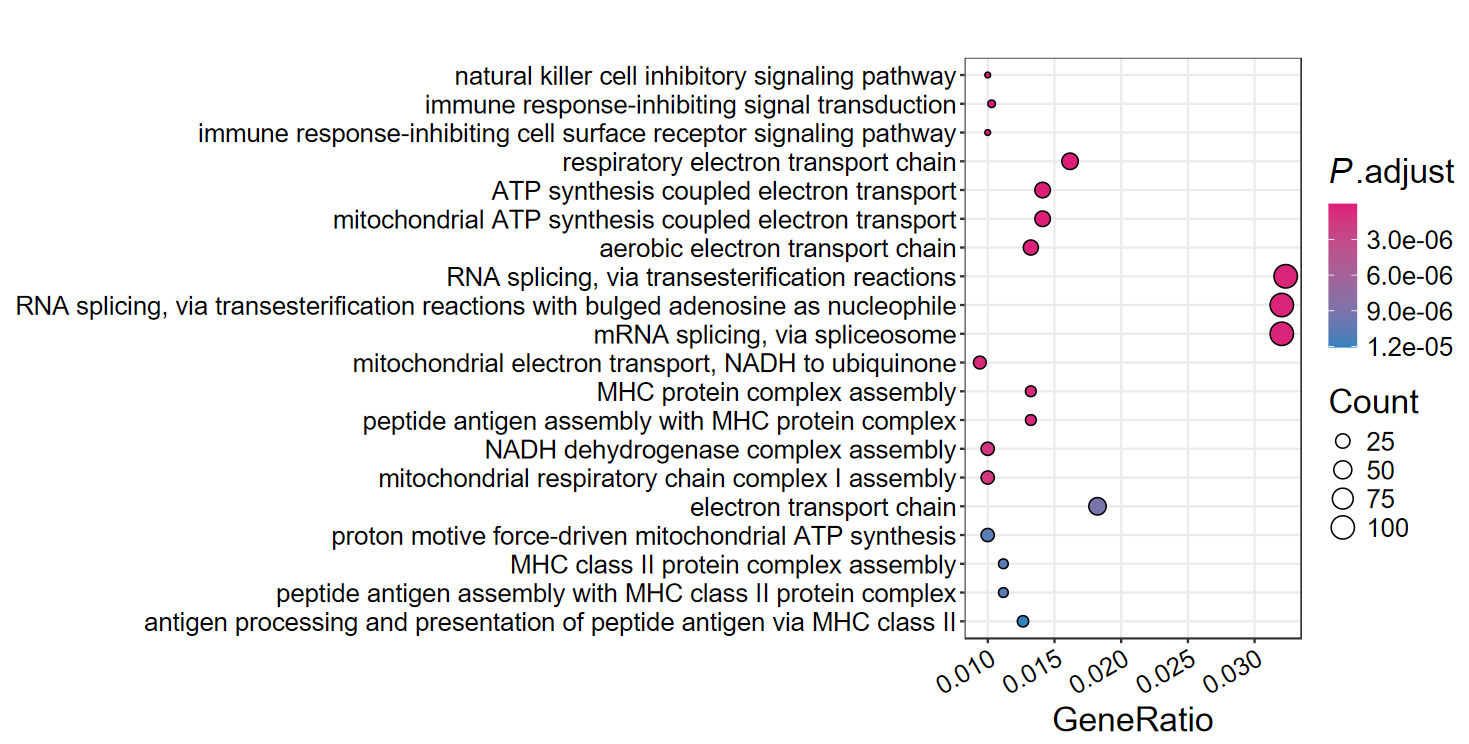
dotplot2(edo, out_name = "DO_dotplot")
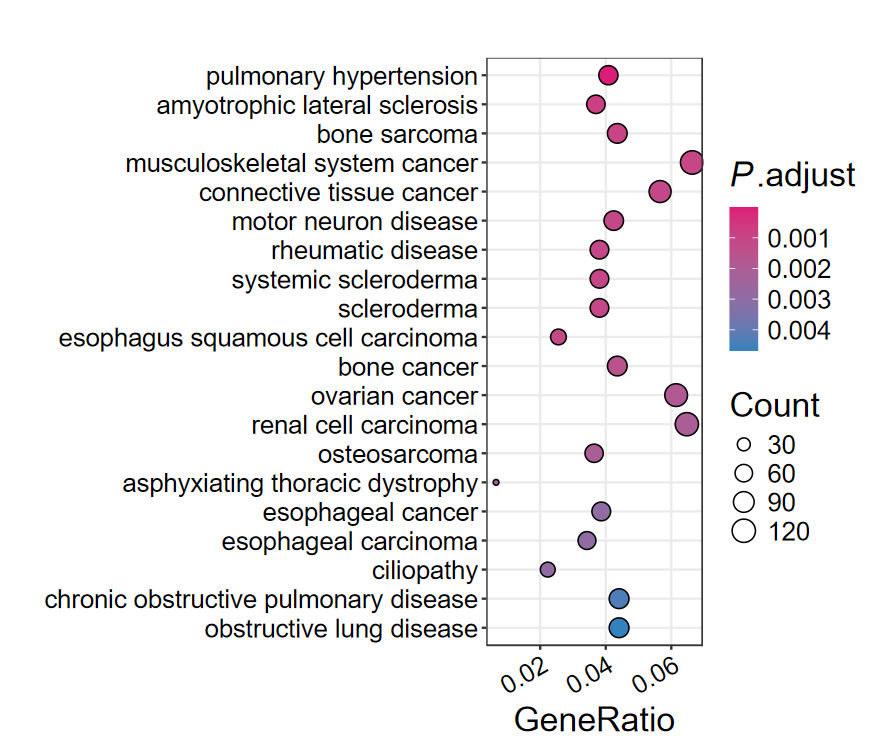
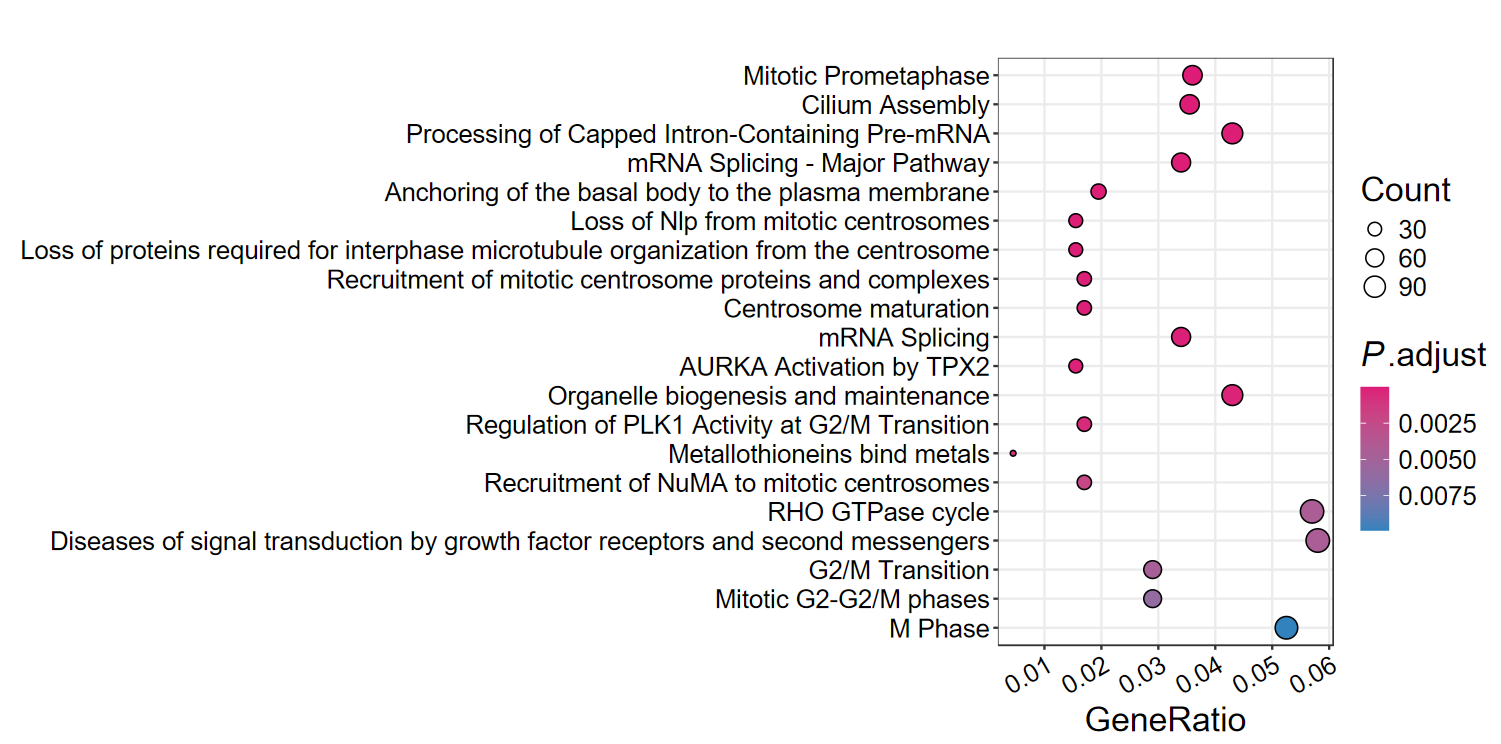
dotplot2(eReactome, out_name = "Reactome_dotplot")
- 同样也支持字符省略
dotplot2(ego, out_name = "GO_dotplot_omit", omit = TRUE)
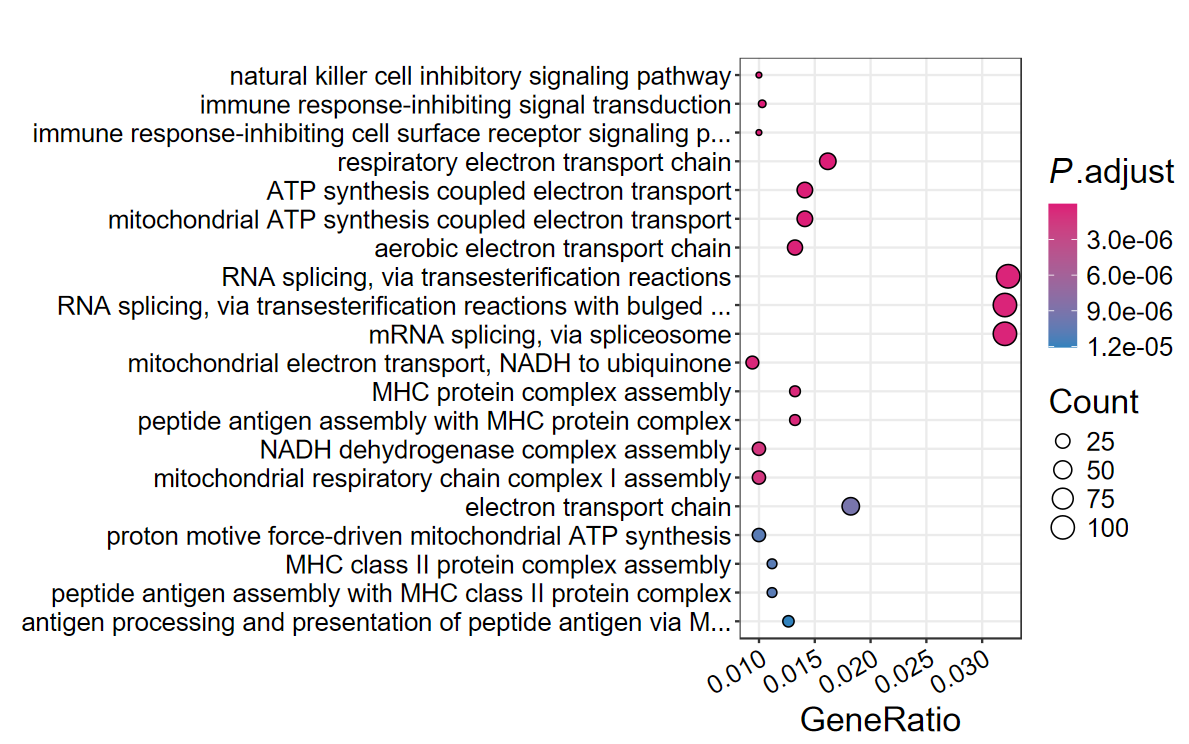
- 同样也支持高亮感兴趣的通路
highlight_paths = c("Non-alcoholic fatty liver disease",
"TNF signaling pathway",
"Alzheimer disease",
"Oxidative phosphorylation"
)
dotplot2(
ekegg,
out_name = "KEGG_dotplot_2",
highlight = highlight_paths,
highlight_color = "red"
)
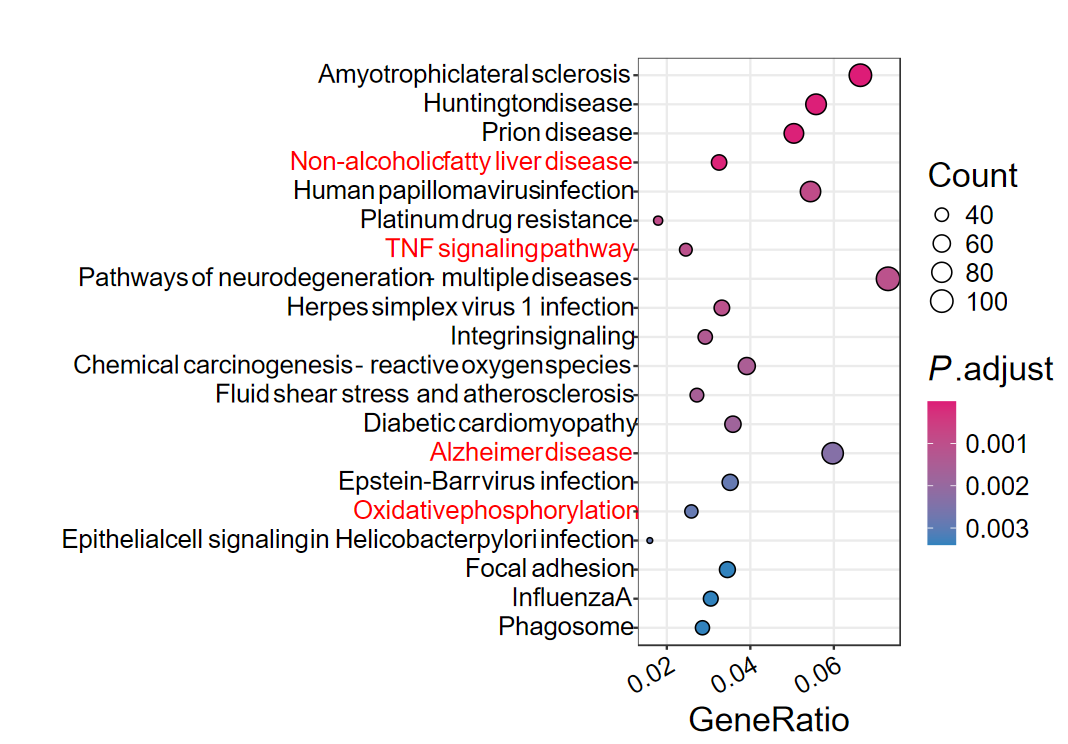
barplot2: 通用条形图
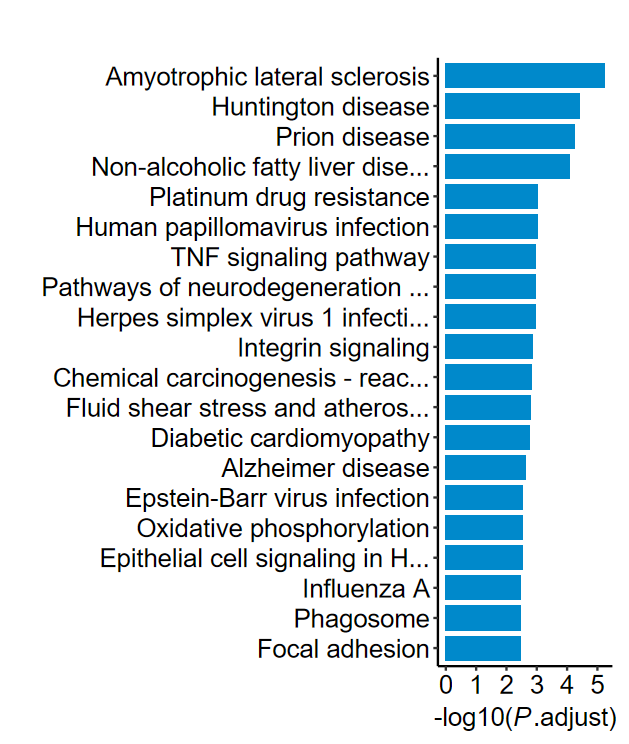
barplot2(ekegg, out_name = "KEGG_barplot")
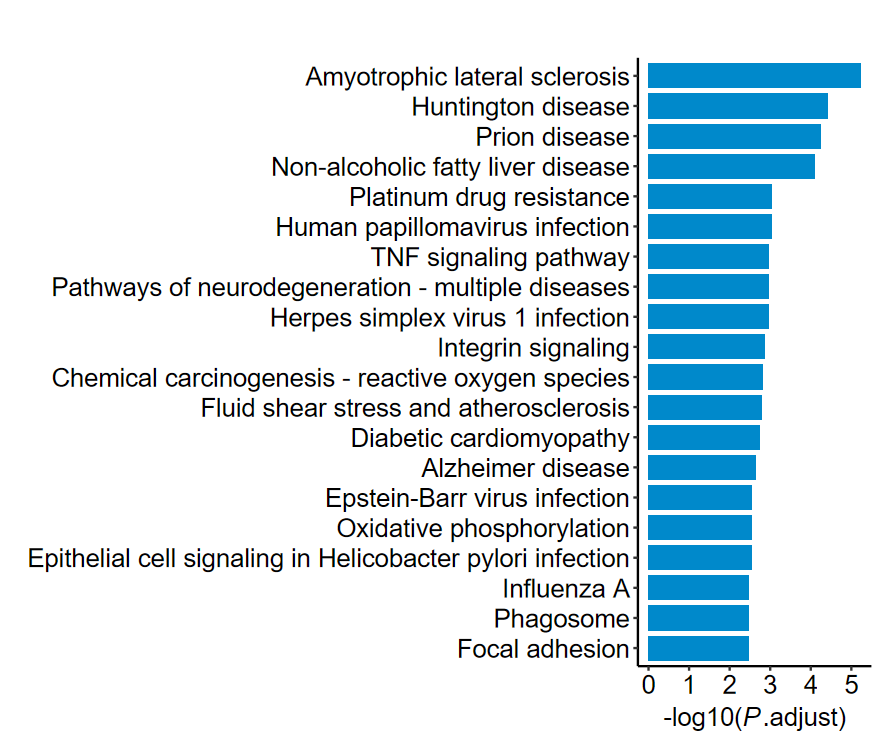
- 增加个数
barplot2(ekegg, out_name = "KEGG_barplot_n30", n = 30)
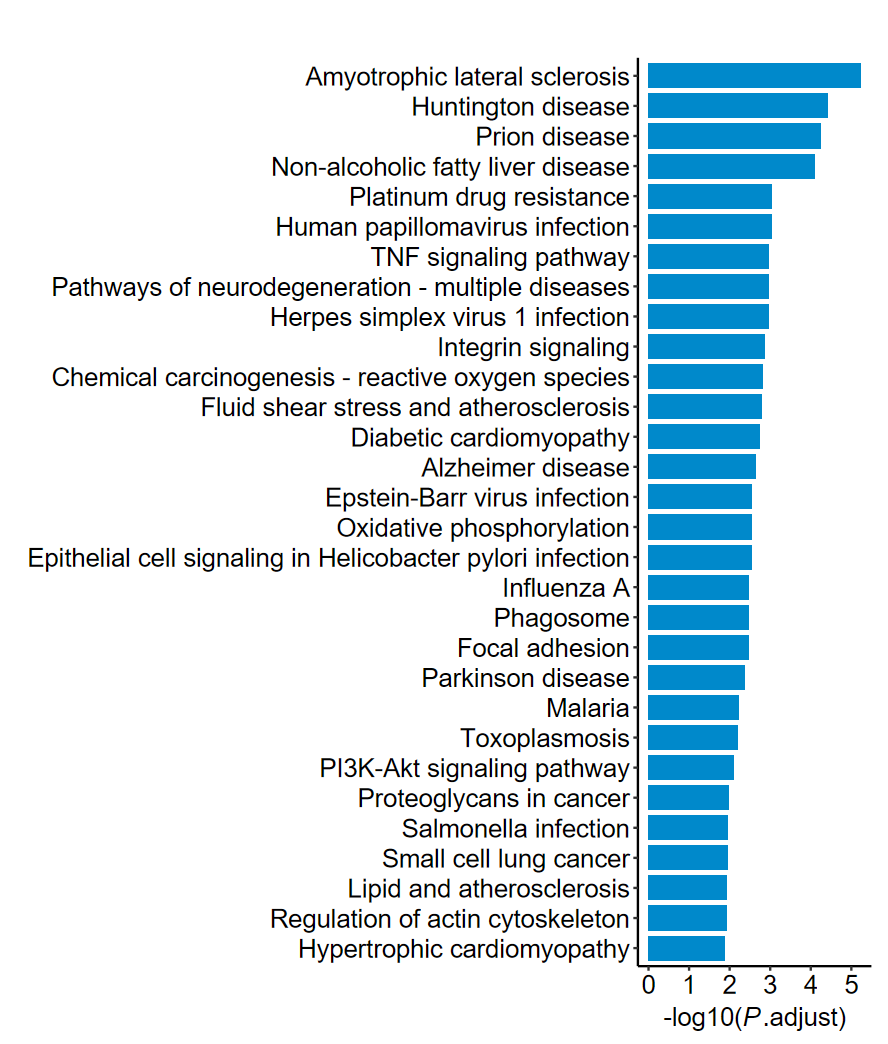
- omit
barplot2(ekegg, out_name = "KEGG_barplot_omit", omit = TRUE, max_omit = 30)
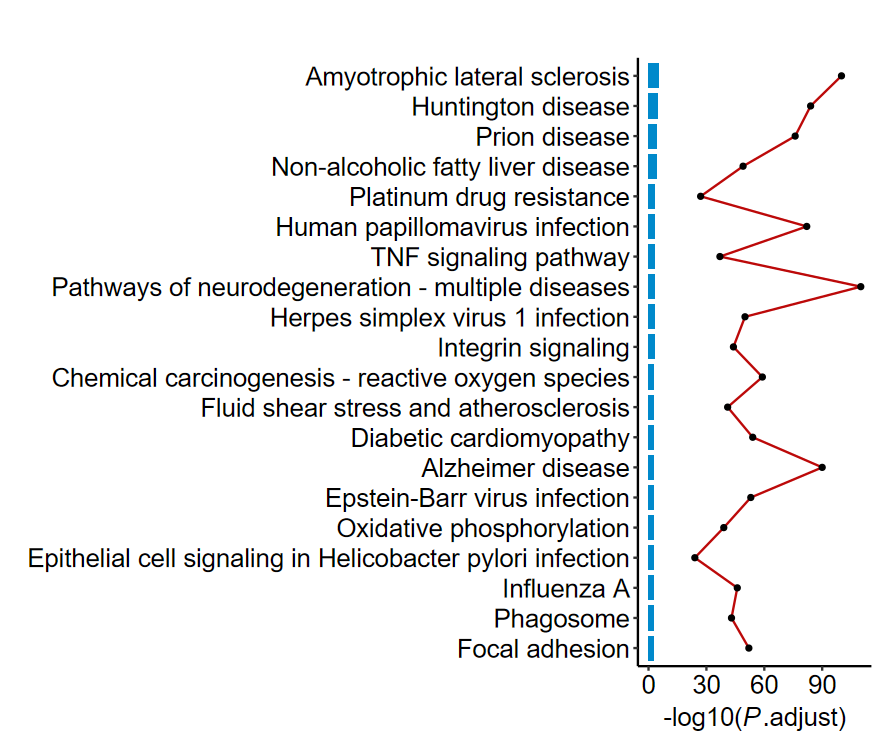
- 还可以添加
add_count_line = TRUE参数增加基因个数 - 但当输入基因比较多的时候, 这条线会很靠右, 所以下面还会优化
barplot2(ekegg, out_name = "KEGG_barplot2", add_count_line = TRUE)
Warning message:
"[1m[22mUsing `size` aesthetic for lines was deprecated in ggplot2 3.4.0.
[36mℹ[39m Please use `linewidth` instead."
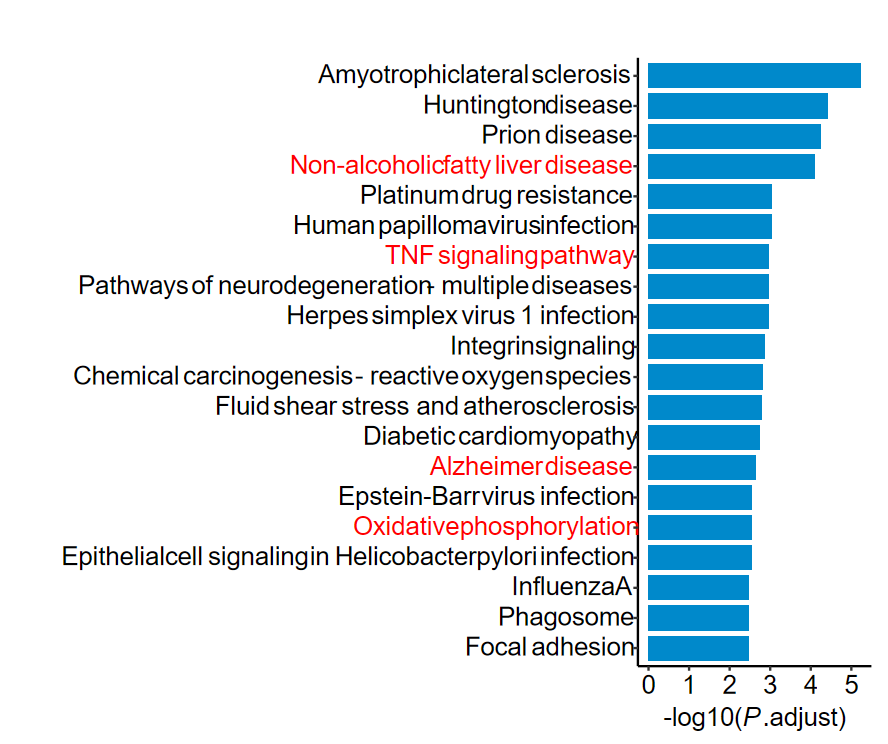
- 同样也支持高亮感兴趣的通路
highlight_paths = c("Non-alcoholic fatty liver disease",
"TNF signaling pathway",
"Alzheimer disease",
"Oxidative phosphorylation"
)
barplot2(
ekegg,
out_name = "KEGG_barplot_3",
highlight = highlight_paths,
highlight_color = "red"
)
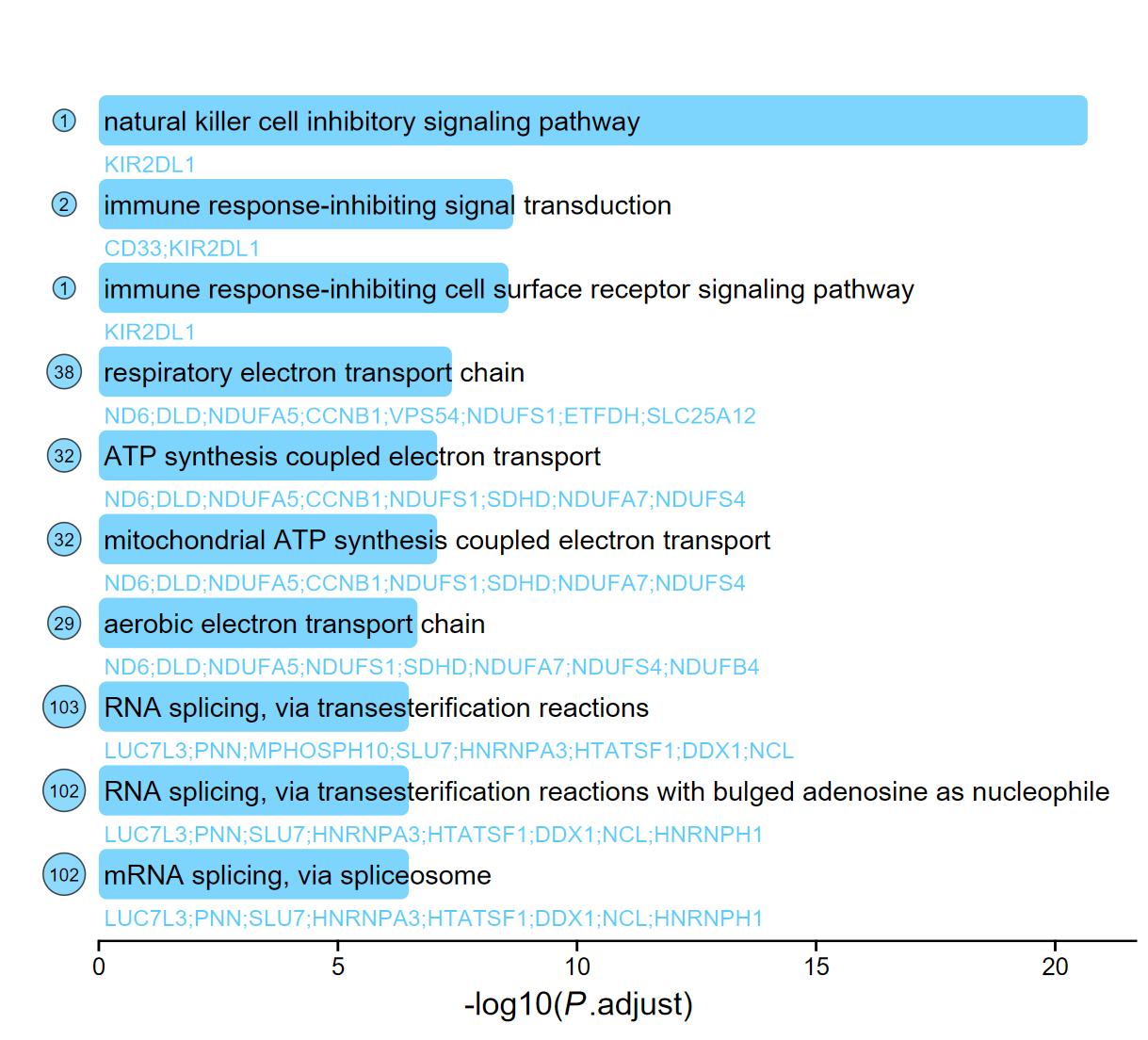
barplot_fancy: 条形图进阶版¶
- 条形图带圆角
- 添加基因名
- 基因数量放在左侧
- 去除图例, 整体即简洁美观, 又信息丰富
barplot_fancy(ego, out_name = "barplot_fancy")
- 增加个数, 也是自动适配宽高
p <- barplot_fancy(ego, out_name = "barplot_fancy_n15", n = 15)
p

p <- barplot_fancy(ego, out_name = "barplot_fancy_n20", n = 20)
p

- 省略字符
barplot_fancy(ego, out_name = "barplot_fancy_omit", omit = TRUE)
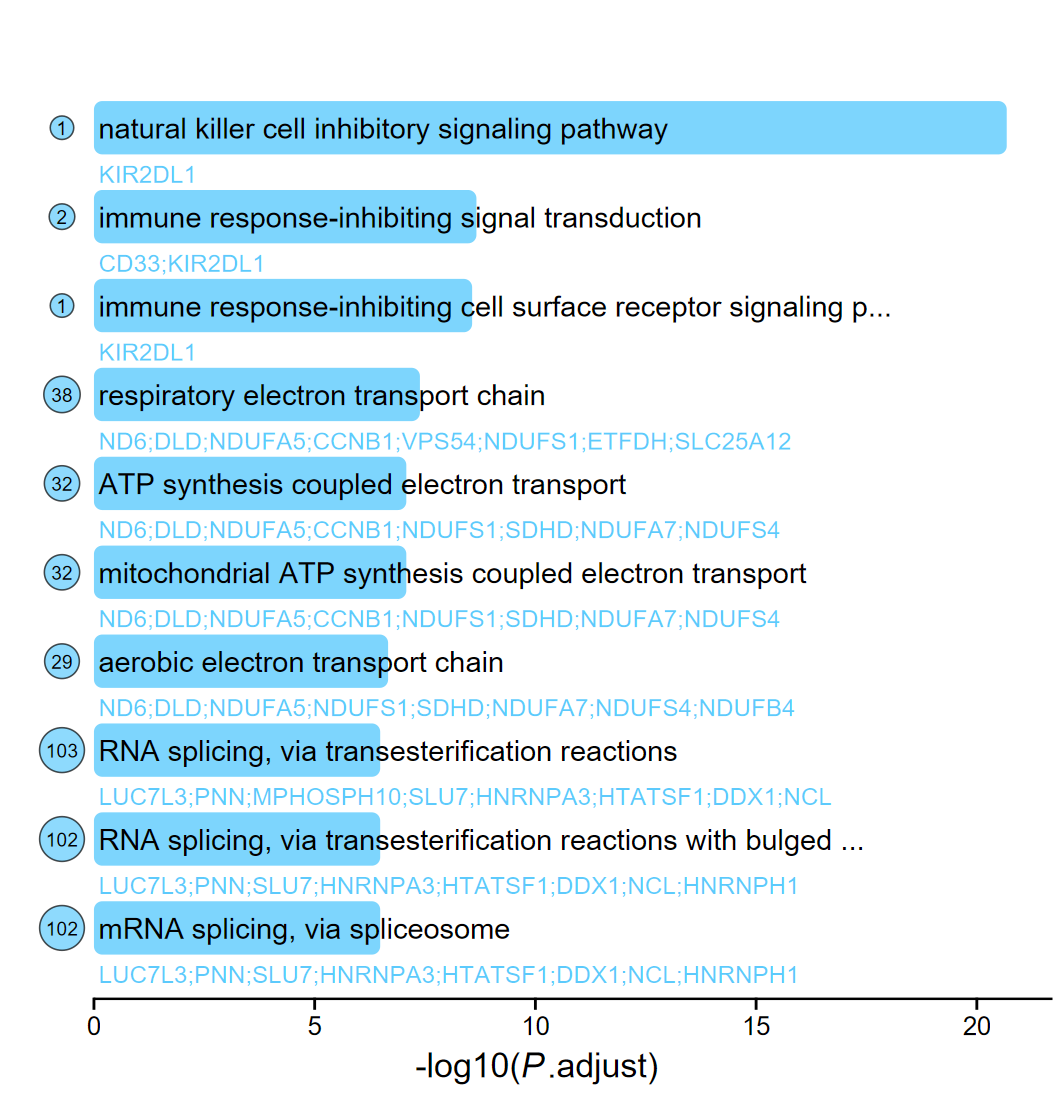
- 换个颜色
barplot_fancy(ekegg, out_name = "barplot_fancy_omit_2", omit = TRUE, fill = "#B368FC")

barplot_fancy(
ego,
out_name = "barplot_fancy_omit_title",
omit = TRUE,
fill = "#B368FC",
title = "GO enrichment",
ylab = "C1: EndoMT COL1A1+ EC"
)
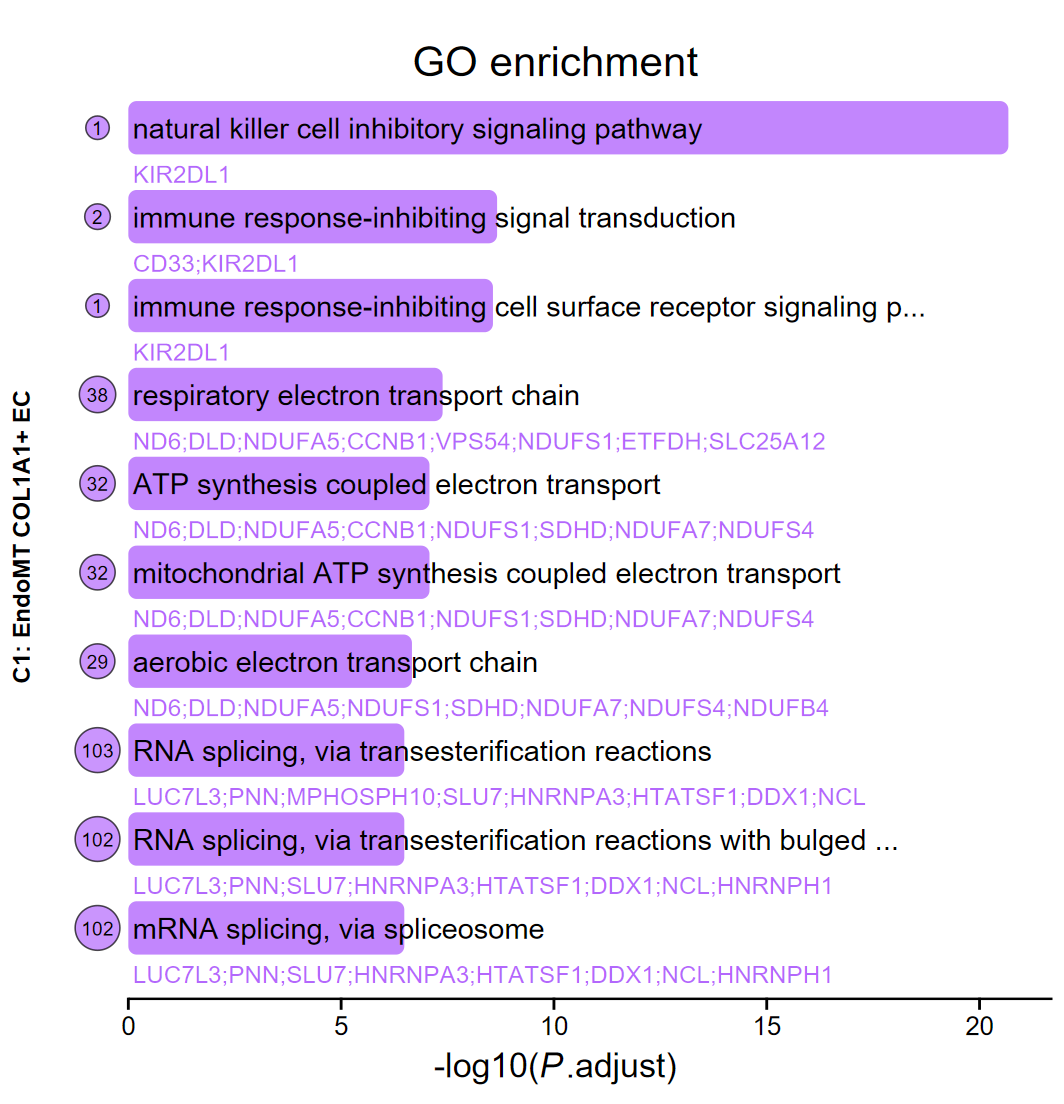
- 不同颜色放在一起看起来很好看
p1 <- barplot_fancy(ego, out_name = "barplot_fancy_omit", omit = TRUE, title = "GO")
p2 <- barplot_fancy(ekegg, out_name = "barplot_fancy_omit", omit = TRUE, fill = "#B368FC", title = "KEGG")
set_image(15, 10)
p1 + p2
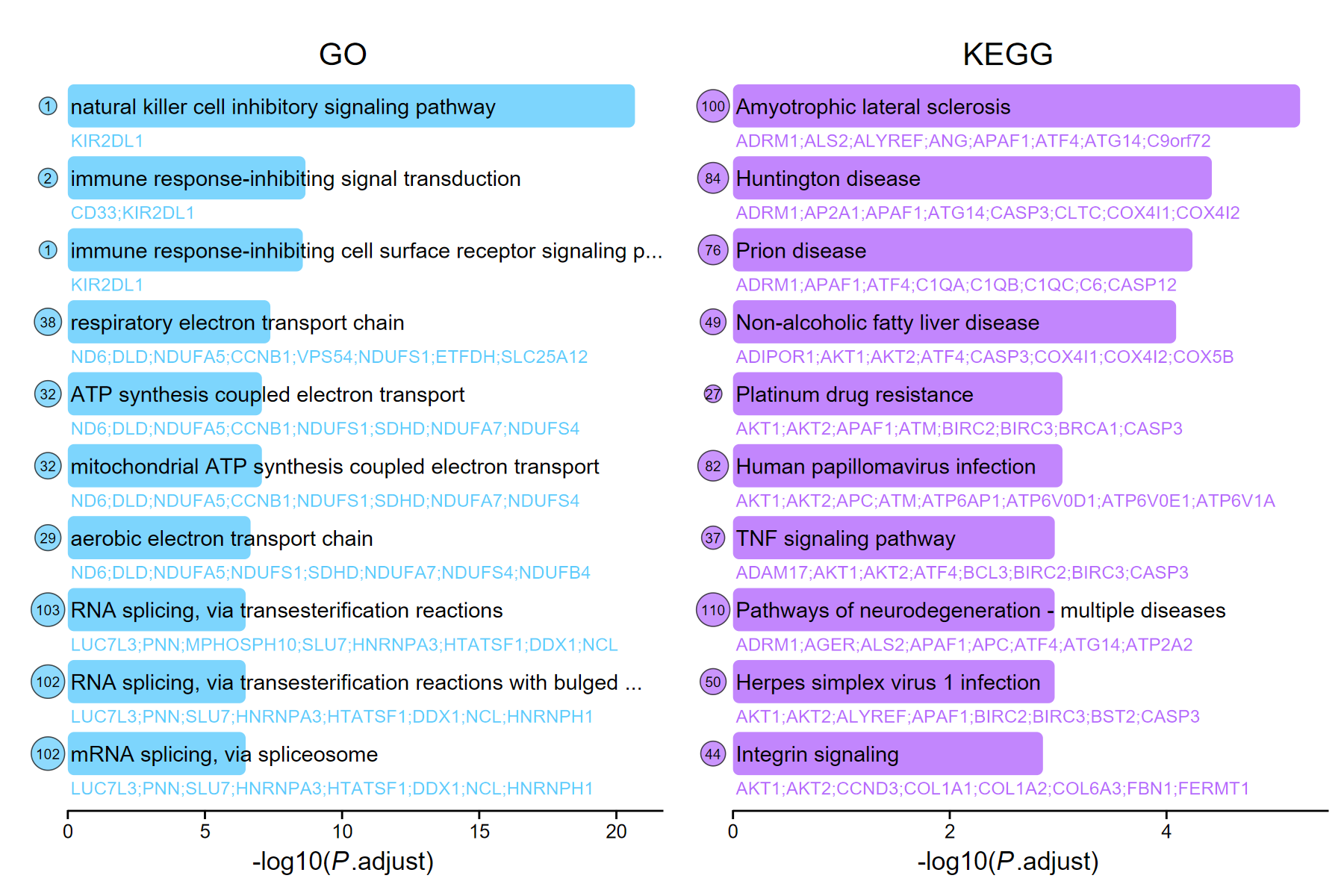
barplot_fancy_category: 标注多种类型的富集结果
- 需要准备含有多种ONTOLOGY数据框格式富集结果
enrich_list = c(split(ego@result, f = ego@result$ONTOLOGY), list(ekegg@result, edo@result, eReactome@result))
names(enrich_list) = c("GO: BP", "GO: CC", "GO: MF", "KEGG", "DO", "Reactome")
shared_cols = Reduce(intersect, lapply(enrich_list, colnames))
enrich_merge = Reduce(rbind, lapply(enrich_list, function(x) x[, shared_cols]))
enrich_merge$ONTOLOGY = rep2(names(enrich_list), sapply(enrich_list, nrow))
enrich_merge$ONTOLOGY = factor(enrich_merge$ONTOLOGY, levels = names(enrich_list))
hd(enrich_merge)
table(enrich_merge$ONTOLOGY)
Type: data.frame Dim: 925 × 10
ID Description GeneRatio BgRatio pvalue
GO:0002769 GO:0002769 natural killer cell inhibitory signaling pathway 34/3402 36/21288 3.281964e-25
GO:0002765 GO:0002765 immune response-inhibiting signal transduction 35/3402 63/21288 6.761106e-13
GO:0002767 GO:0002767 immune response-inhibiting cell surface receptor signaling pathway 34/3402 61/21288 1.268932e-12
GO:0022904 GO:0022904 respiratory electron transport chain 55/3402 140/21288 2.577823e-11
GO:0042773 GO:0042773 ATP synthesis coupled electron transport 48/3402 117/21288 7.858191e-11
GO: BP GO: CC GO: MF KEGG DO Reactome
559 93 75 57 100 41
- 可以通过参数
pathways指定想要展示的通路, 最好每种通路都有 - 如果不指定参数
pathways, 则默认展示每种富集结果的前5个
p <- barplot_fancy_category(enrich_merge, out_name = "barplot_fancy_category")
p
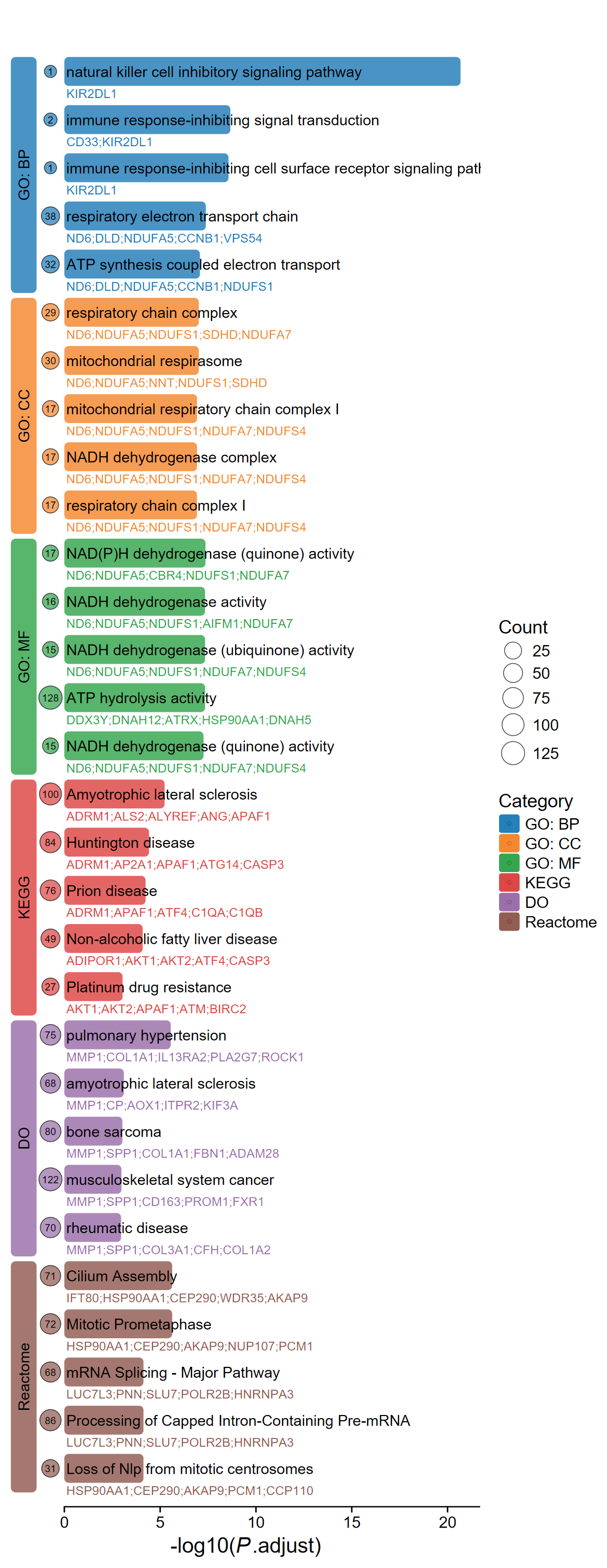
- 设置每个种类的个数
barplot_fancy_category(enrich_merge, out_name = "barplot_fancy_category_2", n_each = 3)

- 指定要展示的通路, 这里随机抽取模拟指定想展示通路
set.seed(1234)
show_paths = sample(enrich_merge$Description, 25)
barplot_fancy_category(enrich_merge, out_name = "barplot_fancy_category_3", pathways = show_paths)
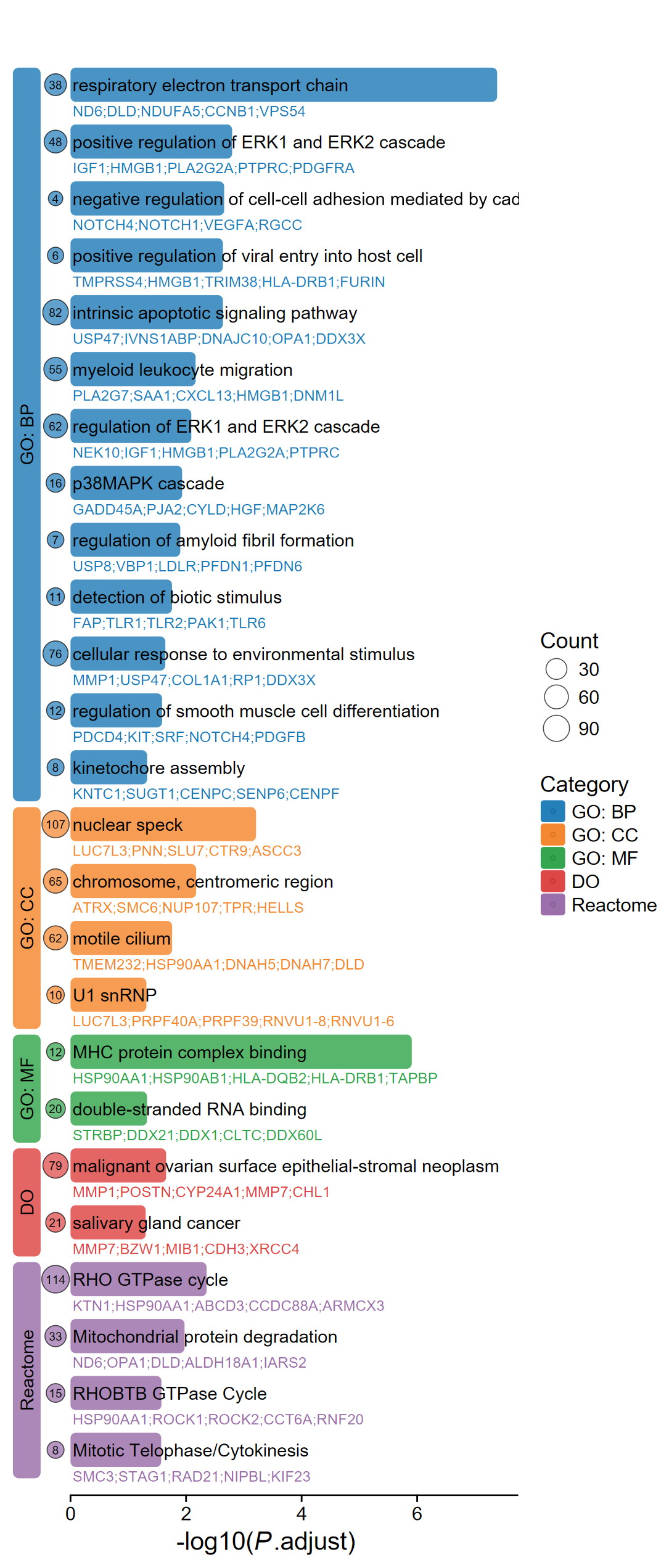
- 由于KEGG占比太少了, 没有被取到, 函数会自动去除该类别
barplot_fancy_GO: 对三种类型的通路进行可视化
- 调用
barplot_fancy_category函数, 继承它的参数
barplot_fancy_GO(ego, out_name = "barplot_fancy_GO")

软件安装
setwd("../")
公共依赖包
-
依赖包自己安装一下好吧, 无非是那几种安装方法
-
install.packages、BiocManager::install、remotes::install_github
-
软件安装问题不免费答疑, 这是基本功, 也跟我的开发关系不大
-
ggplot2 (>= 3.5.2),
-
ggthemes (>= 5.1.0),
-
ggtext (>= 0.1.2),
-
gground(>= 1.0.1), 在github上
-
ggprism(>= 1.0.5),
-
grid (>= 4.4.1),
-
clusterProfiler (>= 4.12.3),
-
org.Hs.eg.db (>= 3.19.1),
-
DOSE (>= 3.30.2),
-
ReactomePA (>=1.48.0)
-
rvest (>= 1.0.4)
自研依赖包
- 当然也依赖我自己开发的一些R包, 在这里提供了压缩包, 直接运行可快速安装
- fanyi2 (>= 1.0.0)
- fastR (>= 1.6.3)
install.packages.local <- function(pkg, dir = "."){
files <- grep(paste0("^", pkg, ".*\\.gz$"), dir("."), value = TRUE)
if(length(files) == 0) stop("未找到匹配的本地包文件")
versions = unname(sapply(basename(files), function(f) strsplit(f, "_|\\.tar")[[1]][2]))
versions_order = order(versions, decreasing = TRUE)
version_latest = versions[versions_order[1]]
latest_file = files[versions_order[1]]
if(length(versions) == 0){
stop("未检索到安装包, 请确认R包压缩包已放置与当前目录, getwd()查看当前目录!")
} else {
message("检索到版本: ", paste(versions, collapse = "、"))
}
if(requireNamespace(pkg, quietly = TRUE)){
version_installed = packageVersion(pkg)
if(version_installed >= version_latest){
message("已安装最新版本: ", version_latest, ", 无需安装!")
} else {
message("当前安装版本: ", version_installed, ", 将安装最新版本: ", version_latest, "...")
install.packages(latest_file, repos = NULL, type = "source", upgrade = "never", force = TRUE)
}
} else {
message("未安装此R包, 将安装最新版本: ", version_latest, "...")
install.packages(latest_file, repos = NULL, type = "source", upgrade = "never", force = TRUE)
}
}
# 安装 fastR
install.packages.local("fastR")
检索到版本: 1.5.0、1.6.3
已安装最新版本: 1.6.3, 无需安装!
# 安装 fanyi2
install.packages.local("fanyi2")
检索到版本: 1.0.0
已安装最新版本: 1.0.0, 无需安装!

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐


 11
11 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)