告别“刻舟求剑”,PackDock如何搞定蛋白质的“七十二变”?
PackDock 结合生成式 AI 与物理先验攻克蛋白受体柔性对接难题,现已入驻 SciMiner 平台,提供即开即用的云端侧链建模与构象分析体验。
在基于结构的药物设计(SBDD)中,分子对接(Molecular Docking) 就像是给一把“锁”(蛋白质口袋)找一把合适的“钥匙”(药物分子)。然而,这把“锁”并不是静止不动的,它会呼吸、会变形。长期以来,如何处理蛋白质的柔性(Flexibility) 一直是计算化学领域的痛点。
近日,中国科学院上海药物研究所郑明月团队提出蛋白–配体复合物柔性结构建模新方法 PackDock。该方法将生成式 AI 与物理算法相结合,用于预测柔性蛋白–配体对接构象,并在多种应用场景中展现出良好的精度与效率,同时具备较强的泛化能力。相关研究成果以 “Flexible protein–ligand docking with diffusion-based side-chain packing” 为题,于 2025 年 12 月 24 日在 Proceedings of the National Academy of Sciences of the United States of America(美国国家科学院院刊)在线发表。

01 “刻舟求剑”的刚性对接
最传统的分子对接,我们称之为刚性对接(Rigid Docking)。
在这个模型里,我们将蛋白质视为一块坚硬的岩石,它的形状完全固定不变;而药物分子则像是一块可以在空间中旋转、平移的磁铁。算法的任务就是把药物塞进蛋白质的口袋里,看哪里卡得最紧、能量最低。
-
优点:计算速度极快,适合大规模虚拟筛选。
-
缺点:“刻舟求剑”。实际上,蛋白质是由氨基酸组成的柔性长链,口袋里的氨基酸侧链是可以随着可旋转键转动的。如果晶体结构中的侧链位置稍微挡了一点路,刚性对接就会认为“撞车”了(Clash),从而把一个潜在的好药误判为无效。
这就是为什么当我们拿Apo结构(无配体结合的空蛋白)去对接时,成功率往往很低——因为口袋还没“张开”。
02 “大海捞针”的传统柔性对接
为了解决这个问题,科学家提出了柔性对接(Flexible Docking)。
传统的柔性对接(如 AutoDock Vina 的柔性模式)允许口袋周围的氨基酸侧链发生旋转。这听起来很美好,但在计算上却是一个组合爆炸的噩梦。
-
挑战:假设口袋里有10个氨基酸,每个氨基酸有3种可能的旋转角度,那么就有3^10种组合。
-
做法:传统算法通常使用启发式搜索(如遗传算法)或旋转异构体库(Rotamer Library)来不断尝试。
-
痛点:“大海捞针”。搜索空间太大,导致计算非常慢;而且往往容易卡在局部最优解,算出一些奇奇怪怪、不符合物理化学规律的构象。
03 破局者:PackDock 方法
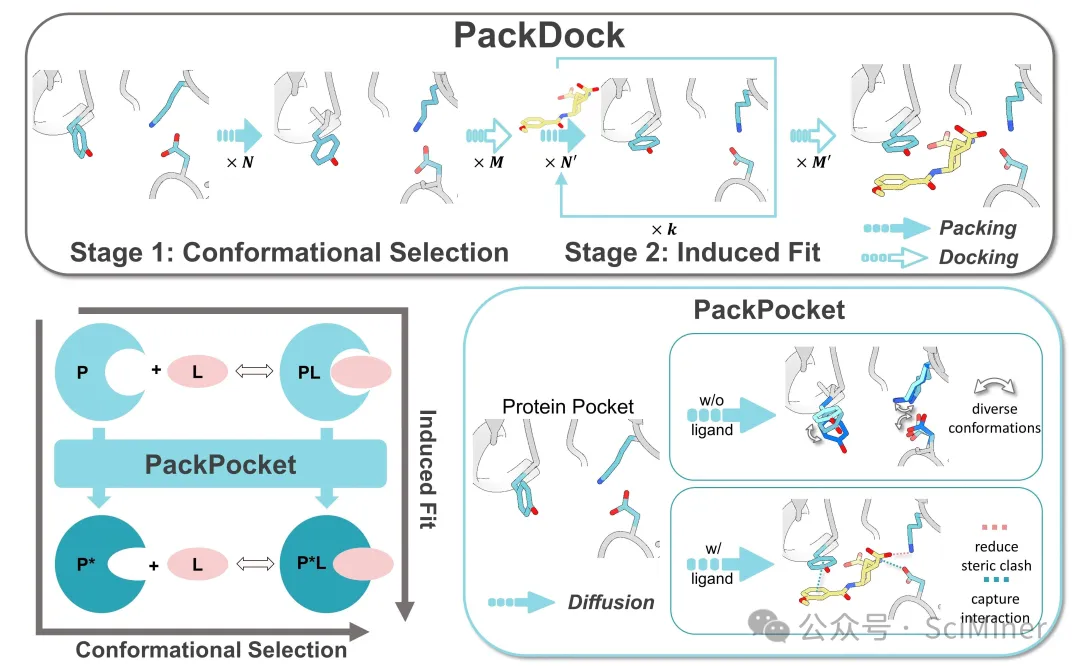
图1. PackDock 方法示意图。a:PackDock 工作流程;b:PackPocket 对侧链构象分布的建模;c:等变图神经网络预测侧链扭转角。
为了既能处理柔性,又不陷入计算黑洞,PackDock 应运而生。
它的核心思想是:不要去盲目“搜索”侧链的位置,而是用AI直接“生成”合理的侧链构象。
PackDock 引入了一个名为 PackPocket 的核心模块,这是一个基于扩散模型(Diffusion Model) 的侧链预测模型。它不像传统方法那样去穷举角度,而是学习了大量蛋白质结构的物理规律,能够直接“画”出符合能量最低原理的侧链位置。
PackDock 的两大理论基石
生物学上,小分子结合蛋白质主要有两种机制,PackDock 完美复刻了这两步:
1. 构象选择(Conformational Selection):蛋白质本身就在不断“呼吸”,在多种构象间跳跃。
2. 诱导契合(Induced Fit):当小分子结合后,蛋白质会进一步调整形状来“抱紧”小分子。
04 PackDock 是如何工作的?
为了让大家更直观地理解 PackDock 的对接全过程,我们可以把它比作“试鞋”的过程。
第一阶段:构象选择(模拟“脚”的自然舒展)
-
传统刚性对接:相当于鞋店只给你看一只固定形状的鞋(晶体结构),不管你的脚胖瘦,塞不进去就算不合适。
-
PackDock 第一步:
-
输入:拿到一个只有骨架的蛋白质(比如Apo结构)。
-
生成:PackDock 利用扩散模型,预测出口袋在没有配体情况下的多种可能形态。就像是让这只鞋展示它自然状态下的几种“撑开”或“收缩”的样子。
-
初筛:把药物分子(脚)分别试着塞进这些不同形态的口袋(鞋)里,进行初步对接。这一步利用了蛋白质天然的动态变化,总有一款形态能让药物大概放进去。
-
第二阶段:诱导契合(模拟“鞋”的贴合调整)
-
传统方法:脚塞进去后,鞋就不动了。
-
PackDock 第二步:
1. 条件生成:现在药物分子(脚)已经大概在口袋里了。PackDock 再次启动扩散模型,但这次它是看着药物的位置来生成侧链的。
2. 微调:模型会调整周围氨基酸侧链的角度,让它们主动去避开药物的原子,或者形成更好的氢键/疏水作用。就像是一双高科技的智能鞋,感知到脚的位置后,自动调整内衬的形状来完美包裹住脚。
3. 重对接:在调整好的完美口袋里,再次精细调整药物的位置,得到最终结果。
05 应用验证:前瞻性虚拟筛选实验
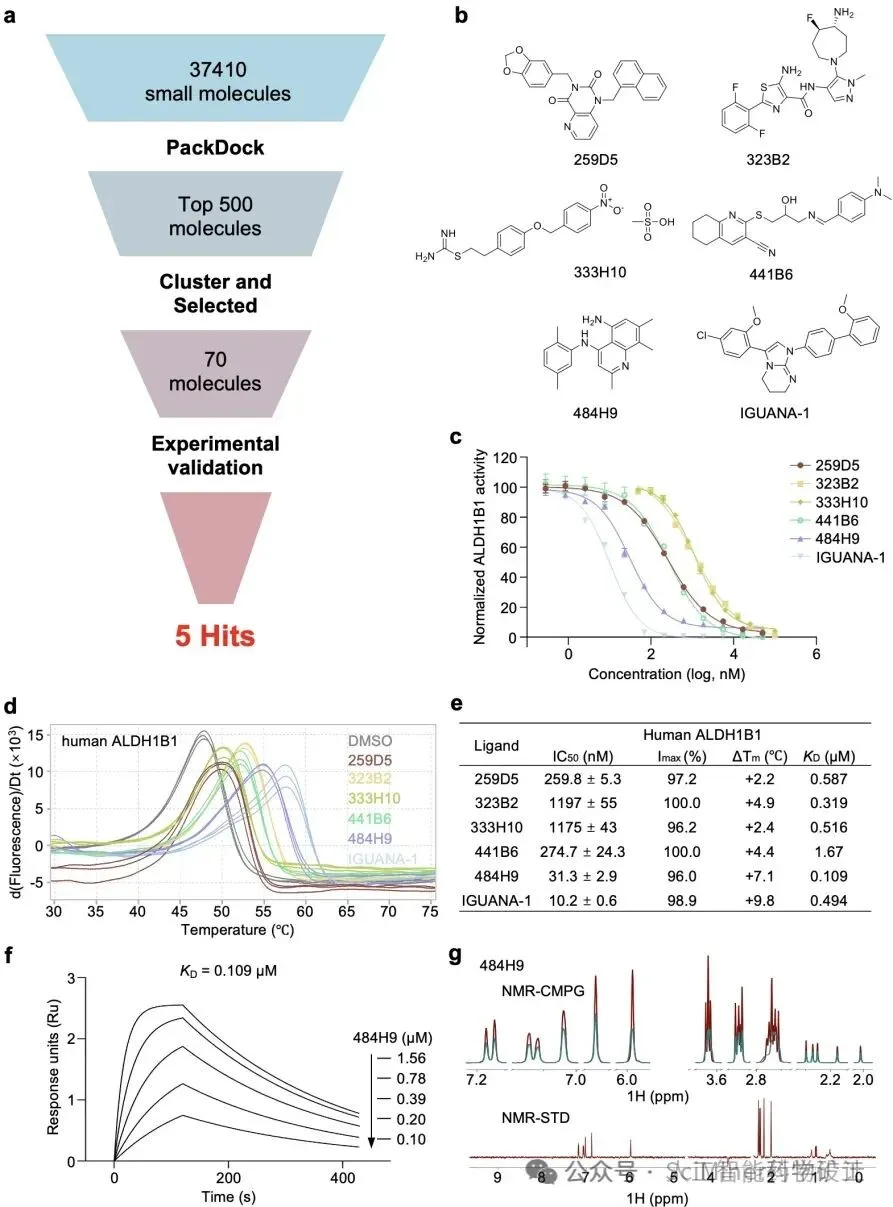
图2. PackDock 筛选 ALDH1B1 的新骨架化合物。a:筛选流程示意图;b:筛选得到的 hit 分子与参考分子 IGUANA-1 的化学结构;c:5 个 hit 分子及参考分子的剂量–反应曲线;d:hit 分子与参考分子对 ALDH1B1 蛋白热稳定性的影响;e:实验活性鉴定结果汇总;f:SPR 测定 484H9 与 ALDH1B1 的结合亲和力;g:484H9 与 ALDH1B1 的核磁共振实验。
为了评估 PackDock 在实际药物发现流程中的应用能力,研究团队以 ALDH1B1 为靶点开展了一项前瞻性虚拟筛选实验,如图2所示。通过使用 PackDock 对内部小分子库进行筛选,并对优选化合物进行多层级实验验证,最终获得 5 个具有新型骨架的 ALDH1B1 抑制剂,其中 1 个化合物表现出纳摩尔级的结合亲和力。该结果表明,PackDock 不仅在基准任务上具备优势,也能够在真实筛选中产出具有实际价值的命中分子,体现了其面向药物发现应用的潜力。
06 总结与展望
PackDock 的巧妙之处在于,它用生成式AI替代了昂贵的搜索算法。
1. 速度快:不需要遍历无数种侧链组合,扩散模型直接生成最可能的几种构象。
2. 精度高:在 Cross-Docking(交叉对接)测试中,PackDock 在使用 Apo 结构(空蛋白)对接时,成功率显著高于传统柔性对接方法,甚至接近了使用 Holo 结构(结合态蛋白)进行刚性对接的“天花板”水平。
3. 意义:这对于新药研发非常重要,因为大多数新靶点我们只有空蛋白的结构(或者AlphaFold预测的结构)。PackDock 让我们可以更自信地针对这些难成药的靶点进行虚拟筛选。
PackDock 正式入驻 SciMiner 平台
为了让这一前沿算法更广泛地服务于生物医药研究者,PackDock 已正式部署至 SciMiner 药物发现云平台,并基于分子活性信息构造合成数据,持续优化更新模型。
● 即开即用的云端算力:无需本地安装与维护,登录平台即可快速启动 PackDock 任务,让复杂的柔性建模过程触手可及。
● 便捷柔性对接:直接调用 PackPocket 模块建模侧链柔性,解决 apo 或折叠结构在对接时口袋不匹配的痛点,获取高置信度的复合物构象。
● 直观结果分析与可视化:对接生成的构象支持在线三维预览,方便研究者直接观察、评估配体与口袋侧链的相互作用模式。
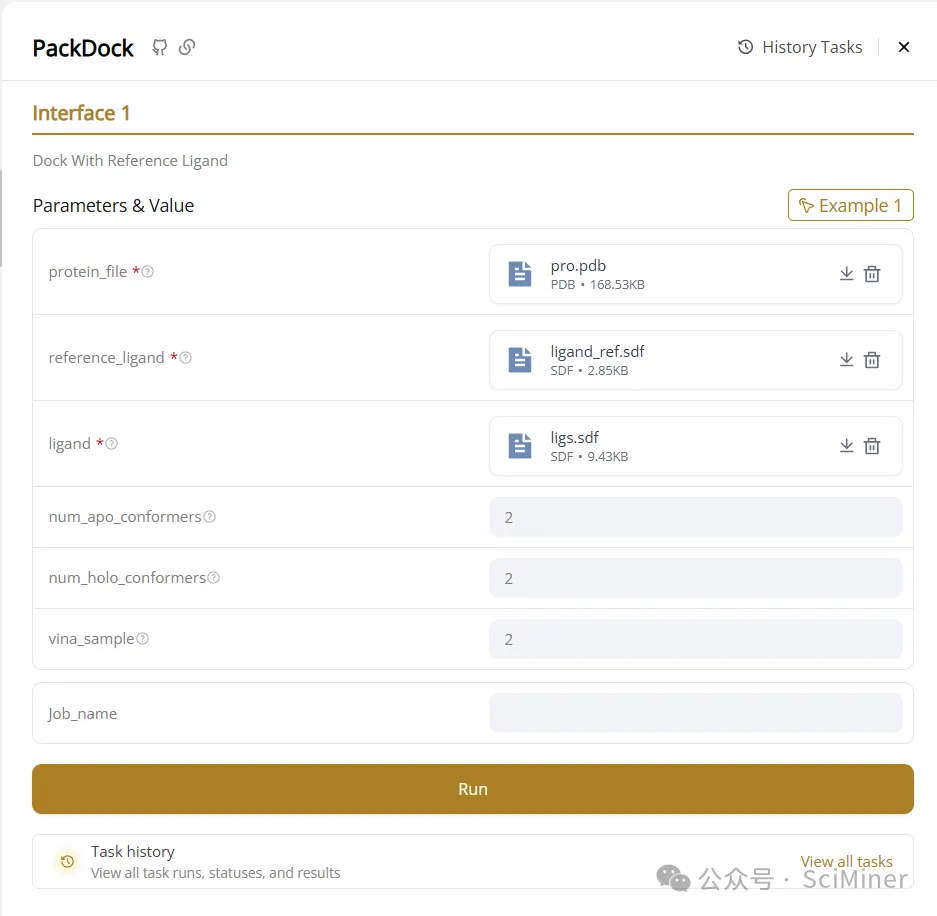
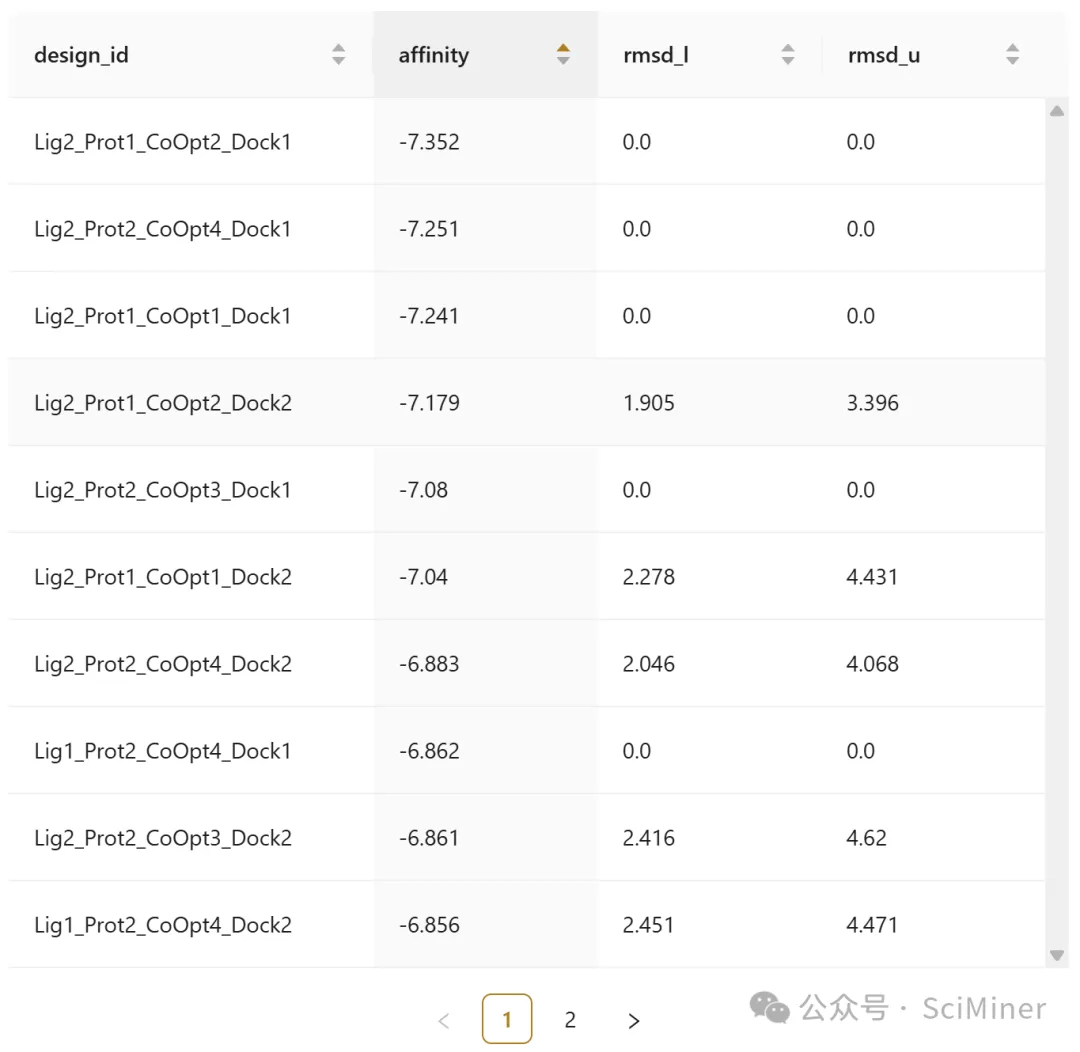
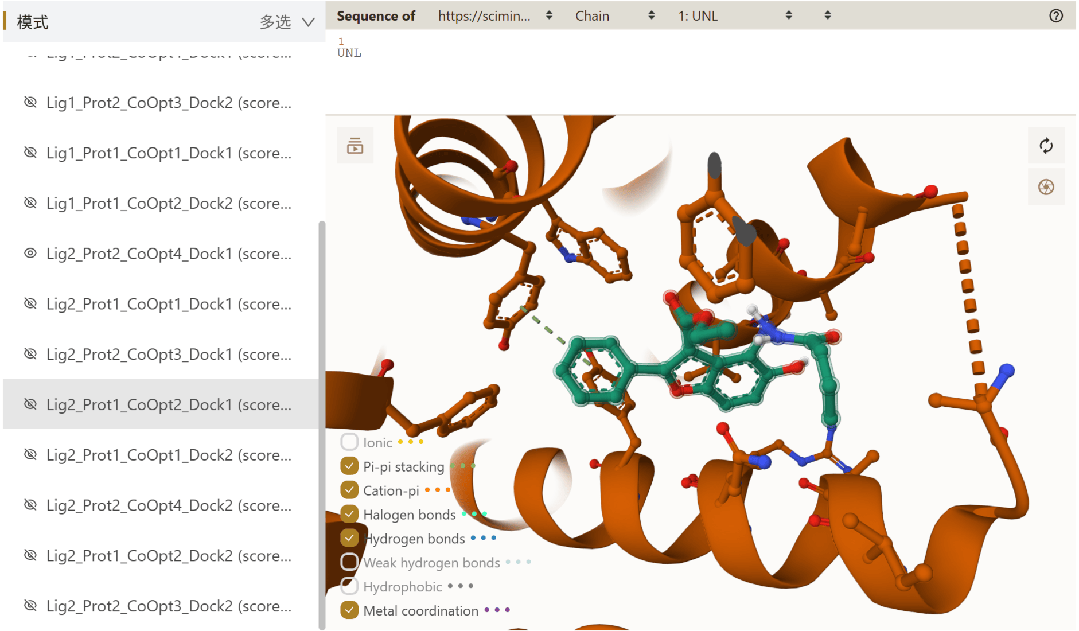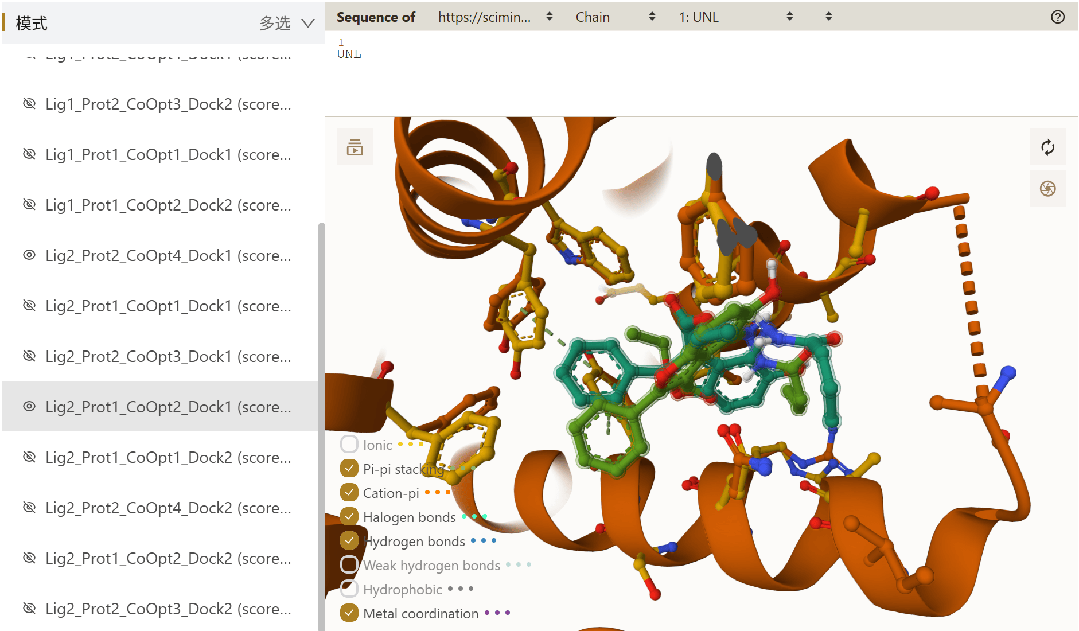
图4. PackDock 输出结果,可以看到侧链的柔性
PackDock 的上线进一步丰富了 SciMiner 在基于结构的药物设计(SBDD)领域的技术储备。我们诚邀广大科研同仁登录 SciMiner 平台,亲自体验这款融合物理先验与深度学习的柔性对接利器,加速新药研发的发现进程。
● 立即体验:https://sciminer.tech/utility?tool=PackDock
● 结果示例链接(免登录查看):https://sciminer.tech/share?id=50cf0f20-cb02-4c19-9d17-6248f21dadc3&type=API_TOOL

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐


 8
8 0
0- 0



 已为社区贡献4条内容
已为社区贡献4条内容

所有评论(0)