听说你的DNA大模型缺乏训练语料——OMG宏基因组数据集
研究团队整合全球两大宏基因组资源(IMG与MGnify),构建了OpenMetaGenomic(OMG)语料库,包含3.1万亿碱基对和33亿蛋白编码序列。针对宏基因组数据分散、质量参差等问题,提出严格质量控制流程,并开发首个混合模态基因组语言模型gLM2,能同时学习蛋白编码区与基因间隔区特征。实验表明该模型在核酸任务和蛋白相互作用预测中表现优异,为基因组基础模型研究提供了新范式。数据集、模型和代码
和人类语料相比,DNA序列大模型的语料要匮乏很多,同时也是因为人类无法直读的缘故,使得DNA大模型的训练语料尤为珍贵。

今天我们介绍来自 Tatta Bio、美国能源部联合基因组研究所(JGI)以及欧洲生物信息学研究所(EMBL-EBI) 的联合研究团队,在计算机科学顶级会议 ICLR 2025 上发表的题为 “The OMG dataset: An open metagenomic corpus for mixed-modality genomic language modeling” 的研究论文。该研究系统整合了全球规模最大的两大宏基因组资源——IMG 与 MGnify,构建了一个总规模达 3.1 万亿碱基对、包含 33 亿蛋白编码序列 的开放宏基因组语料库 Open MetaGenomic(OMG)。
工作背景:
宏基因组来源的微生物基因组蕴含着极高的分子多样性,覆盖了生命之树中大量尚未被探索的进化分支。然而,宏基因组数据天然具有非结构化、质量参差和高度冗余等特征,使其长期未能被充分用于生物语言模型训练。 具体而言,宏基因组序列面临三方面核心挑战:
-
一、数据分散于不同数据库,缺乏统一、可直接下载的整合语料;
-
二、预处理成本高,原始拼装序列需经过复杂的基因预测与严格的质量控制;
-
三、数据存在显著采样偏倚与重复问题,由于序列长度不一且缺乏可靠的分类学标签,传统基于聚类或分类的去重与平衡方法难以适用。
这些因素共同限制了宏基因组在大规模生物语言模型中的应用潜力。
核心创新点:
1. 大规模、可训练的混合模态宏基因组语料库(OMG)
数据来源与规模
OMG 语料库整合了全球规模最大的两大宏基因组资源——JGI 的 IMG 与 EMBL-EBI 的 MGnify,总规模达 3.1 万亿碱基对,包含 33 亿条蛋白编码序列(coding sequences, CDS),显著扩展了可用于基因组语言模型训练的序列多样性。
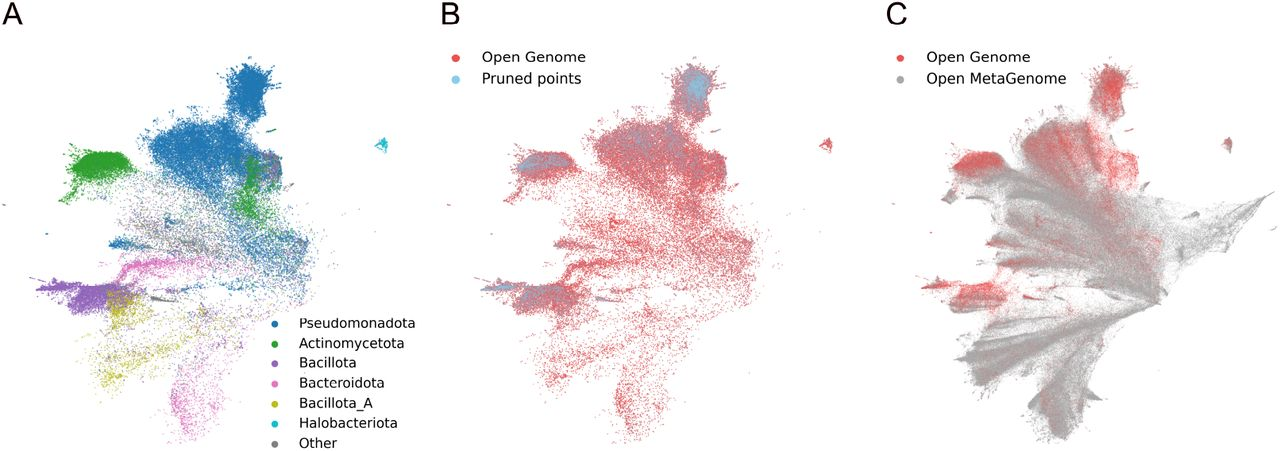
混合模态组织方式
OMG 以真实宏基因组 contig 为基本单位,对序列进行混合模态表示:其中蛋白编码区以氨基酸序列(AA)表示,基因间隔区以核酸序列(NA)表示,并严格保留基因顺序与上下游关系,从而维持真实的基因组上下文结构。
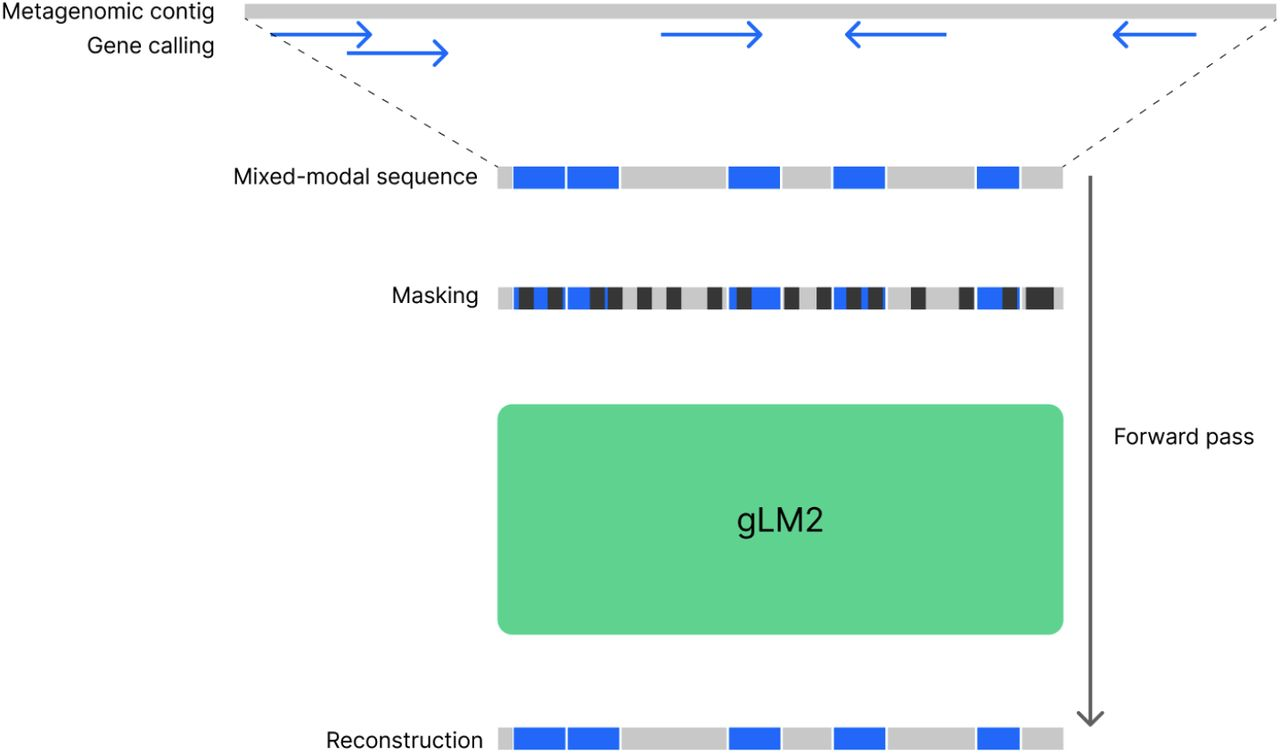
将宏基因组重叠群的基因预处理成混合模态序列,该序列由 CDS 元件(蓝色)和 IGS 元件(灰色)组成。然后对该混合模态序列进行 30% 的掩蔽处理,并使用掩蔽标记重建目标训练 gLM2 模型。
这比传统只用蛋白或短片段序列更能捕获真实生物语义。
2. 面向宏基因组的系统性质量控制与预处理流程
针对宏基因组序列天然存在的拼装错误、片段化和噪声问题,研究团队构建了一套严格且可扩展的质量控制流程。
具体而言,预处理步骤包括:
-
去除过短 contig
-
剔除边缘基因预测不完整的编码区
-
过滤高比例不确定碱基(N/X)的低质量区域
-
排除异常超长的蛋白编码区和基因间隔区。
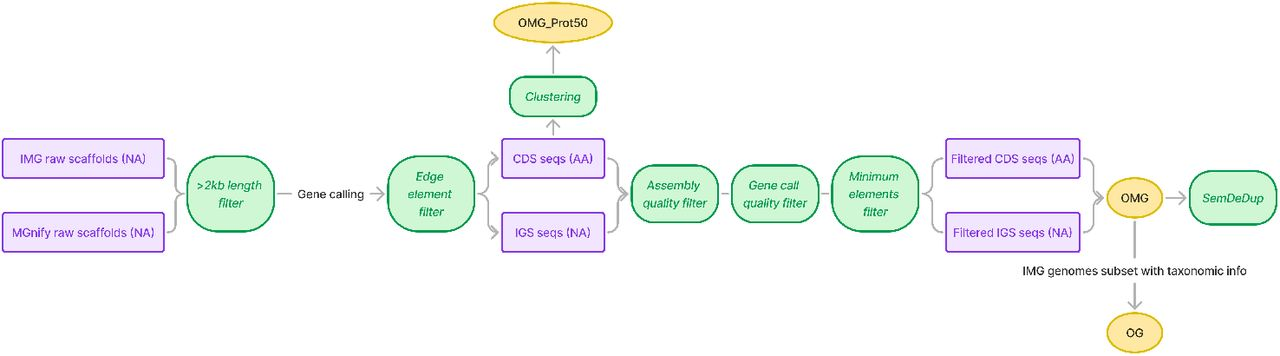
序列(紫色)经过过滤步骤(绿色)后,生成了本文提供的三个 Hugging Face 数据集(黄色)。“
通过上述步骤,显著降低了宏基因组数据中的噪声,为后续模型训练提供了高质量输入。
3. 首个gLM2框架的混合模态基因组语言模型
基于 OMG 语料库,研究团队提出并训练了首个gLM2框架的混合模态基因组语言模型 。该模型以真实宏基因组 contig 为输入单元,统一建模蛋白编码区(氨基酸序列)与基因间隔区(核酸序列),并显式编码基因顺序与链方向信息。
通过混合模态输入与掩码语言建模目标,gLM2 能够在单一模型框架内同时学习蛋白功能特征与非编码调控序列特征,为跨模态基因组理解提供了统一表示空间。
训练效果与性能评估
在模型性能展示方面,模型训练效果的展示相对克制,更侧重于验证混合模态数据设计的有效性。主要有以下几点
1. 在核酸任务上的表现对比
尽管 gLM2 的训练语料中只有一小部分是 DNA(核酸)序列,但在这些核酸任务上,gLM2 的表现与 Nucleotide Transformers 相似
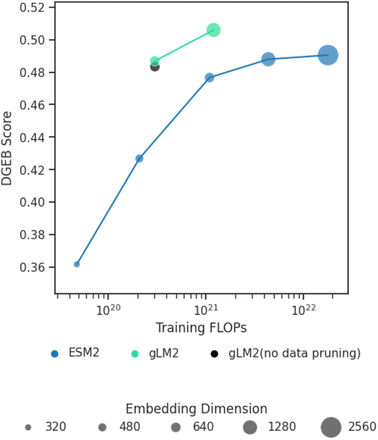
这说明混合模态训练并未削弱模型对核酸序列的学习能力。基因组上下文信息提供了足够的信号,使模型在 NA 任务中仍能保持较强性能
2. 蛋白–蛋白相互作用与进化信号学习
除了定量评估,文章还通过 无监督学习进化信号的案例验证 了 gLM2 的潜力: 模型能够学习蛋白家族之间的协同进化信号,识别蛋白–蛋白接触界面。与仅基于单蛋白序列的模型(如 ESM2 或 Evo)比较,gLM2 在某些复合体接触预测中显示出了更一致的模式这一点从功能和结构一致性上支持了模型能够以无监督方式学习更高阶生物学关系。
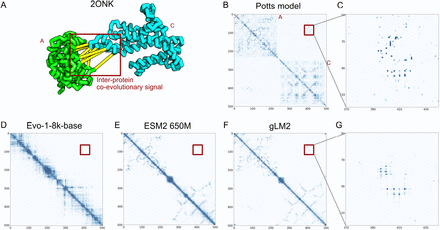
gLM2 学习 2ONK (ModAC) 复合物中的蛋白质-蛋白质界面共进化信号
总结与展望
-
开源数据
目前最大、最规范的开源宏基因组预训练语料库,为社区提供了宝贵的基础设施。
-
混合模态
混合模态基因组建模有望成为基因组基础模型的新范式。 特别是将蛋白编码区与基因间隔区以混合模态形式进行统一建模,能够在单一模型框架内同时捕获蛋白功能信息与基因调控语法。未来,混合模态基因组表示有望取代单一模态的蛋白或核酸建模方式,成为构建通用基因组基础模型的重要方向,并推动模型从“序列理解”迈向“基因组级系统理解”。
-
数据为王
数据中心化与表示结构的设计可能比算法设计更加成为模型性能提升的关键。 相较于单纯依赖模型规模扩展,本研究显示数据构建、质量控制与语义去重等数据中心化策略,对模型性能和泛化能力具有同等甚至更为关键的影响。未来的基因组基础模型研究,或将更加关注数据表示结构、上下文组织方式及跨模态信息整合,从而以更高的计算效率实现更具生物学一致性的模型能力提升。
资源链接:
-
Dataset: https://huggingface.co/datasets/tattabio/OMG
-
Model: https://huggingface.co/tattabio/gLM2_650M
-
Code: https://github.com/TattaBio/OMG
原文出处: Cornman, A., Roux, S., West-Roberts, J., et al. (2025). THE OMG DATASET: AN OPEN METAGENOMIC CORPUS FOR MIXED-MODALITY GENOMIC LANGUAGE MODELING

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐



 21
21 0
0- 0



 已为社区贡献1条内容
已为社区贡献1条内容

所有评论(0)