STREAMS指南:环境及宿主相关微生物组研究中的技术报告标准
STREAMS指南为环境和非人类宿主微生物组研究提供标准化报告框架,包含67项核心条目。该指南在STORMS基础上扩展,涵盖从研究设计到数据分析的全流程,特别针对环境研究的特殊性提供操作建议。通过248名研究人员的共识,STREAMS制定了机器可读的DMP工具模板和8个应用案例,推动微生物组数据的FAIR化。作为持续更新的"活文档",该指南为研究者提供规范化投稿准备,同时为审稿
小编导读
随着微生物组研究从人体健康向环境、植物和动物等领域快速扩展,标准化报告的缺失成为数据共享和重复利用的最大障碍。研究在STORMS人类微生物组指南的基础上,专门为环境和非人类宿主微生物组研究量身定制了STREAMS标准。这套包含67个条目的指南不仅涵盖了从样本采集、实验设计到数据分析的完整流程,还特别针对环境研究的特殊性提供了清晰的操作建议。更贴心的是,研究团队还开发了机器可读的DMP Tool模板和八个不同领域的实战案例,让标准不再是"纸上谈兵"。作为一份会持续更新的"活文档",STREAMS有望成为推动全球微生物组数据FAIR化的重要基础设施,帮助研究者在投稿前就做好规范化准备,也为审稿人提供了统一的评估框架。
研究

微生物组研究的跨学科性质,加上复杂的多组学数据生成,使知识共享变得充满挑战。《加强微生物组研究组织与报告》(STORMS)指南为报告人类微生物组研究的科学手稿中研究信息、实验设计和分析方法的报告提供了清单。在此,在这份共识声明中,我们介绍了环境与宿主相关微生物组研究(STREAMS)指南中技术报告的标准。指南在STORMS基础上扩展,包含67项,支持以标准化且可机器作的方式报告和审查环境(例如陆地、水生、大气和工程)、合成及非人类宿主相关微生物组研究。基于来自28个国家的248名研究人员的意见,我们提供了详细的指导,包括与STORMS的比较,以及展示STREAMS指南应用情况的案例研究。STREAMS和STORMS一样,将成为由联盟更新的活生生社区资源,并结合更广泛社区的共识建设意见。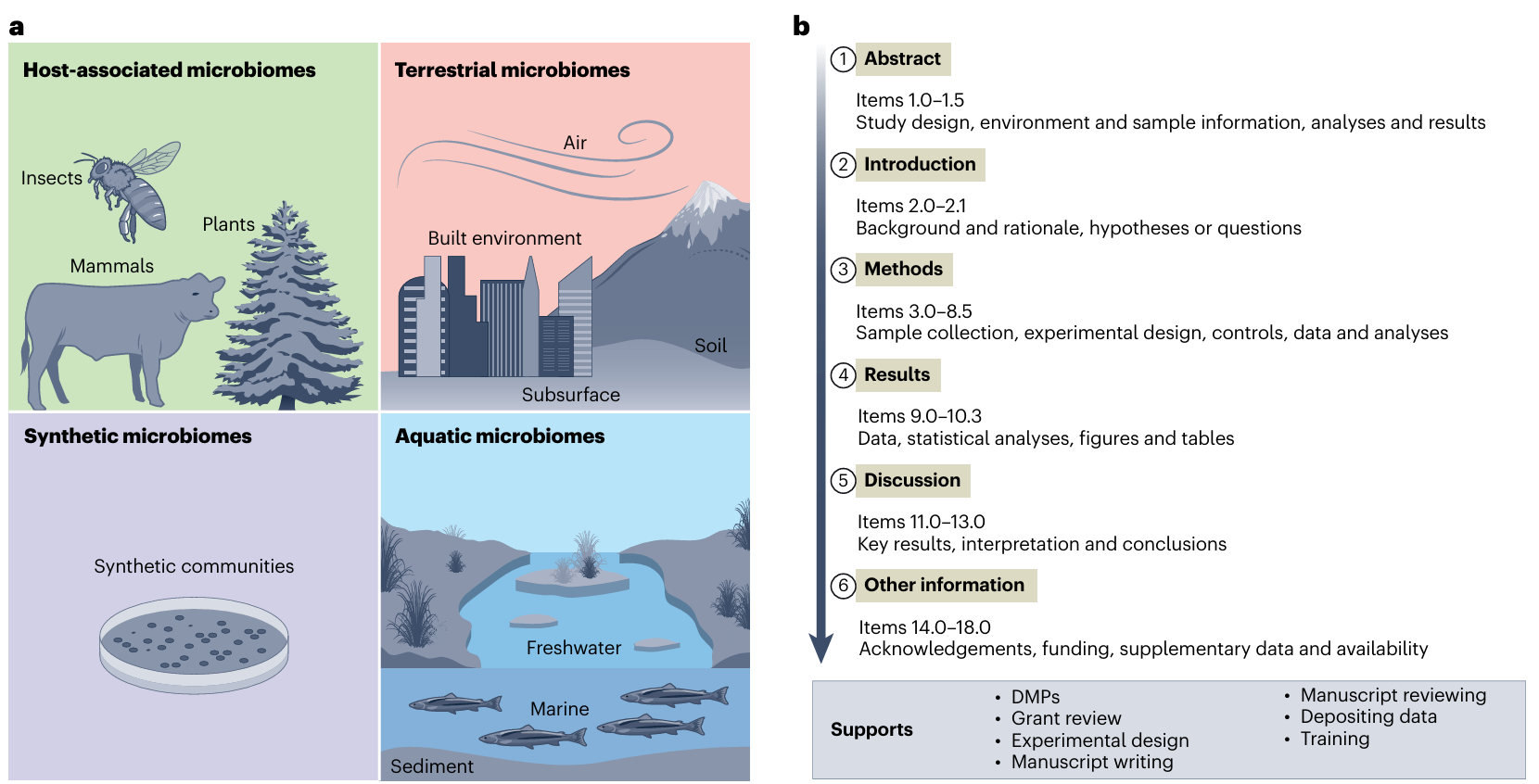
研究人员填写STREAMS指南的8个范例,涵盖涵盖多种样本和数据类型的已发表稿件。
微生物组研究报告指南总结
这份指南为微生物组研究的规范化报告提供了系统性框架,涵盖从研究设计到成果发表的全流程要求。
研究设计与背景阐述
研究者需要在摘要和引言部分清晰阐明研究的科学背景、理论依据和知识空白,明确提出研究假设或科学问题。摘要应采用结构化或非结构化形式,概括研究设计、方法、结果、结论及其科学意义。引言部分需要通过文献综述建立研究的必要性,说明该研究如何填补现有知识缺口。
摘要与引言要求
| 部分 | 条目 | 具体要求 |
|---|---|---|
| 摘要 | 结构化摘要 | 包含背景、方法、结果、结论和意义,可配图形或视频摘要 |
| 研究设计 | 概述研究设计类型 | |
| 环境与样品信息 | 描述微生物组来源、样品类型、特定微生物的学名 | |
| 宿主信息 | 描述宿主分类学特征、健康状态、个体数量 | |
| 实验与组学方法 | 说明实验策略、组学技术、数据类型、是否使用已有数据 | |
| 分析与结果 | 简述分析方法、主要结果及其领域意义 | |
| 引言 | 背景与理论依据 | 总结研究动机、科学证据、理论基础,引用相关文献 |
| 知识缺口 | 明确指出领域内尚未解决的问题 | |
| 假设或研究问题 | 清晰陈述研究问题、目标和假设 |
方法学核心要素
研究设计与样品信息
| 条目 | 具体要求 |
|---|---|
| 研究设计 | 描述整体设计方案,说明是否为已有数据的分析或联合分析 |
| 样品描述 | 使用标准化元数据(如MIxS)详细描述所有样品;合成菌群需说明构建方法和最终组成 |
| 公开数据再利用 | 引用并描述使用的公开样品和数据 |
| 环境背景与地理位置 | 提供环境背景、地理区域、地理坐标(若不提供需说明理由) |
| 相关日期 | 说明是否为纵向/时间序列研究,注明采样频率和关键时间点 |
| 宿主信息 | 描述非人类宿主的分类学信息及相关特征或健康状态 |
| 伦理审查 | 提供许可信息和遵循的伦理准则(如CARE原则) |
| 环境条件与实验处理 | 列出实验处理、环境暴露条件、场地历史(如农业实践、土地利用、气候) |
| 样品采集 | 说明采集方法、是否立即混合样品、具体采样位置 |
| 纳入/排除标准 | 列出环境、宿主或样品的选择标准 |
| 分析样本量 | 解释最终样本量的计算方法,包括对照组数量 |
样品处理与实验操作
| 条目 | 具体要求 |
|---|---|
| 储存与保存 | 描述从采样到实验各阶段的保存方法,包括长期储存方案 |
| 运输 | 说明样品运输或邮寄方式 |
| 提取方法 | 提供核酸、蛋白质、代谢物、脂质等生物分子的提取方法、试剂盒和操作流程 |
| 实验与样品处理 | 描述所有样品修饰或实验操作(如培养、流式分选、同位素标记、表面灭菌、匀浆、过滤、病毒组分纯化、亚采样、密度筛选) |
| 文库制备 | 描述测序文库的制备方法 |
| 去除、富集与多重化 | 详细说明核酸去除或富集方法(如宿主去除、rRNA去除、polyA选择、基于序列的富集、多核酸钳) |
| 引物选择 | 提供引物选择、核酸扩增方法及目标可变区信息 |
质量控制体系
| 条目 | 具体要求 |
|---|---|
| 阳性对照 | 描述使用的阳性对照(如模拟菌群、内标、标准品)及其在实验中的使用阶段;代谢组学和蛋白质组学的标准品 |
| 阴性对照 | 描述使用的阴性对照及其在实验中的使用阶段 |
| 定量与质量评估 | 说明样品质量评估方法、核酸定量方法(分光光度法如NanoDrop或荧光法如Qubit)、RNA质量和核酸片段大小测定方法(如Bioanalyzer)、代谢物和蛋白质的定量与质量信息 |
| 污染控制与识别 | 提供控制或识别环境、试剂、实验室污染的实验室方法 |
| 重复实验 | 描述生物学重复或技术重复的设计,包括重复用于哪些具体步骤 |
| 测序方法 | 描述测序策略(如宏基因组、宏转录组、扩增子测序;长读长或短读长)、测序仪和测序方法/化学试剂 |
| 代谢组学、蛋白质组学及其他组学方法 | 提供方法学和平台信息、样品制备方法、使用的仪器和数据采集方法 |
| 背景数据与关联数据集 | 说明其他生成的数据和元数据的定义、测量或收集方法、应用的转换,提供背景数据或相关数据 |
| 批次效应 | 讨论已知的批次效应来源及其最小化和校正方法 |
数据分析要求
生物信息学分析
| 条目 | 具体要求 |
|---|---|
| 生物信息学分析 | 描述所有生物信息学步骤,包括分类学分析、功能分析或其他分析;描述使用的代码、软件、工作流程和工具 |
| 质量控制 | 描述识别或过滤污染或低质量序列的方法;报告阳性和阴性对照的结果;报告质量阈值和下游步骤的质量信息(如高质量MAGs的鉴定) |
| 标准化 | 描述定量变量的转换方法(如使用百分比代替计数、标准化、稀疏化、分类) |
| 数据库信息 | 明确所有数据库(分类学数据库、代谢物数据库、肽段数据库)及其版本;提供自定义数据库的生成和使用信息 |
统计分析
| 条目 | 具体要求 |
|---|---|
| 统计方法 | 描述所有统计方法的具体计算、公式、软件、工作流程、工具和代码 |
| 缺失数据 | 解释缺失数据的处理方法 |
| 偏倚与混杂变量 | 讨论样品和数据中的潜在偏倚;讨论可能影响结果和暴露的混杂变量;说明控制的变量及其理由;描述最小化潜在偏倚的方法 |
| 亚组分析 | 描述亚组检验方法,包括亚组分离的理由 |
| 敏感性分析 | 描述任何敏感性分析 |
| 显著性标准 | 说明选择报告结果的标准和显著性阈值 |
数据可及性要求
| 条目 | 具体要求 |
|---|---|
| 元数据访问 | 说明样品元数据(人口统计学、环境条件、其他协变量)的访问途径及其与微生物组数据的匹配方法;说明关联数据集和背景数据的访问途径 |
| 宿主数据访问 | 说明宿主数据(如宿主基因组)的访问途径及其与微生物组数据的匹配方法;描述宿主元数据(包括表型信息)的访问和关联方法 |
| 原始数据访问 | 说明原始数据的访问途径,包括解复用信息 |
| 处理后数据访问 | 说明处理后数据的访问途径,确保在文本和相关文件/链接中清楚标注处理"阶段" |
| 软件与源代码访问 | 引用所有使用的软件、工作流程和代码 |
| 可重复性研究 | 声明他人是否以及如何重现所报告的方法和分析 |
结果报告要求
| 条目 | 具体要求 |
|---|---|
| 样品特征与实验结果 | 总结环境、宿主、样品的特征和相关历史信息(如干旱、火灾)、实验和潜在混杂因素;提供表型分析、培养或其他实验结果 |
| 微生物组测序数据 | 报告微生物组分析的所有适用结果和协变量;包括分类学信息、差异丰度分析或其他分析 |
| 代谢组学、蛋白质组学及其他组学数据 | 报告各类组学数据分析结果;描述组学类型如何与其他组学数据集匹配或整合,以及如何解释这些结果 |
| 统计分析 | 报告统计数据分析的结果及其显著性 |
| 图表与图注 | 提供准确、可及、清晰的图表和图注来展示结果 |
讨论与结论
| 条目 | 具体要求 |
|---|---|
| 关键结果 | 参考研究目标和假设总结关键结果 |
| 结果解释 | 考虑研究目标、多重分析、类似研究结果和数据集比较以及其他相关证据,对结果进行合理解释 |
| 局限性 | 讨论研究的局限性,考虑潜在偏倚来源或不精确性 |
| 外推性 | 讨论研究结果的外推性(外部有效性) |
| 持续与未来工作 | 基于研究发现描述潜在的未来研究或正在进行的研究 |
| 结论 | 陈述本研究得出的总体结论(可能需要单独章节) |
其他必要信息
| 条目 | 具体要求 |
|---|---|
| 致谢 | 致谢对研究有贡献但不符合作者资格的人员,使用CRediT角色(https://credit.niso.org)区分作者贡献与致谢;致谢相关设施或机构(如实验室空间、野外台站、计算基础设施);致谢土著土地使用和伦理研究相关信息 |
| 资助 | 提供资助来源(奖励编号)和资助方在本研究及原始研究(如适用)中的作用 |
| 利益冲突 | 包含利益冲突声明 |
| 补充数据与文件 | 说明补充数据和文件的访问途径及其包含的信息;提供不同文件间信息匹配的键或数据字典 |
| 样品与数据可用性 | 提供关于如何以及在何处访问研究相关所有数据的声明;提供相关样品的访问声明 |
| AI使用 | 描述是否使用AI、使用的程序和版本、具体用途(如文献综述、代码生成、数据分析、稿件起草、翻译辅助);参考出版商和资助机构的具体指南,确保遵循最新的AI使用报告要求 |
关于DMP Tool
DMP Tool是由加州数字图书馆开发的免费在线数据管理计划(Data Management Plan)构建平台,可以自动关联用户所在机构信息、资助机构要求和持久标识符,帮助研究者快速生成符合各类标准的机器可读DMP文档。STREAMS团队在该平台上开发了专用模板,让微生物组研究者能够一键生成包含67项指南要素的标准化数据管理方案。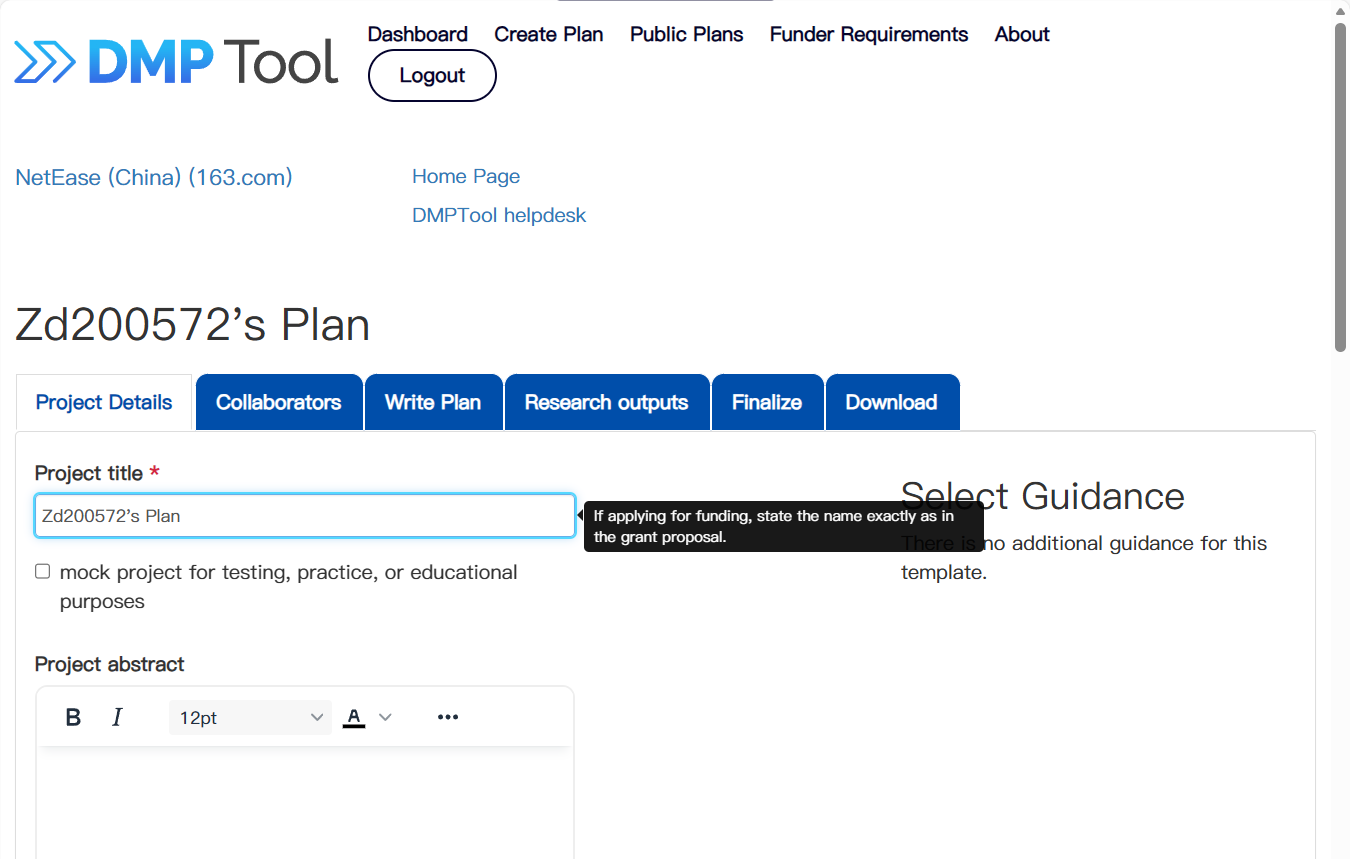
关于STORMS
Strengthening The Organization and Reporting of Microbiome Studies是用于报告人类微生物组研究的清单,分为六个部分,涵盖科学出版物的所有部分,呈现为表格,并留有作者提供意见的空间,并旨在纳入补充材料中。 借鉴了流行病学领域的STROBE指南和基因关联研究的STREGA标准,为人体微生物组研究的手稿撰写提供了系统化的报告框架。该清单已被众多期刊和资助机构广泛采纳,但仅适用于人类相关研究,这也是STREAMS诞生的直接动因——将标准化报告的成功经验拓展到环境和非人类宿主微生物组领域。
STORMS按主要手稿标题列出项目清单。有关每个项目的详细描述及额外指导,请参见STORMS检查表。
3.6项流程图示例。虽然STORMS并非强制要求,但流程图可以帮助可视化最终分析样本的计算过程。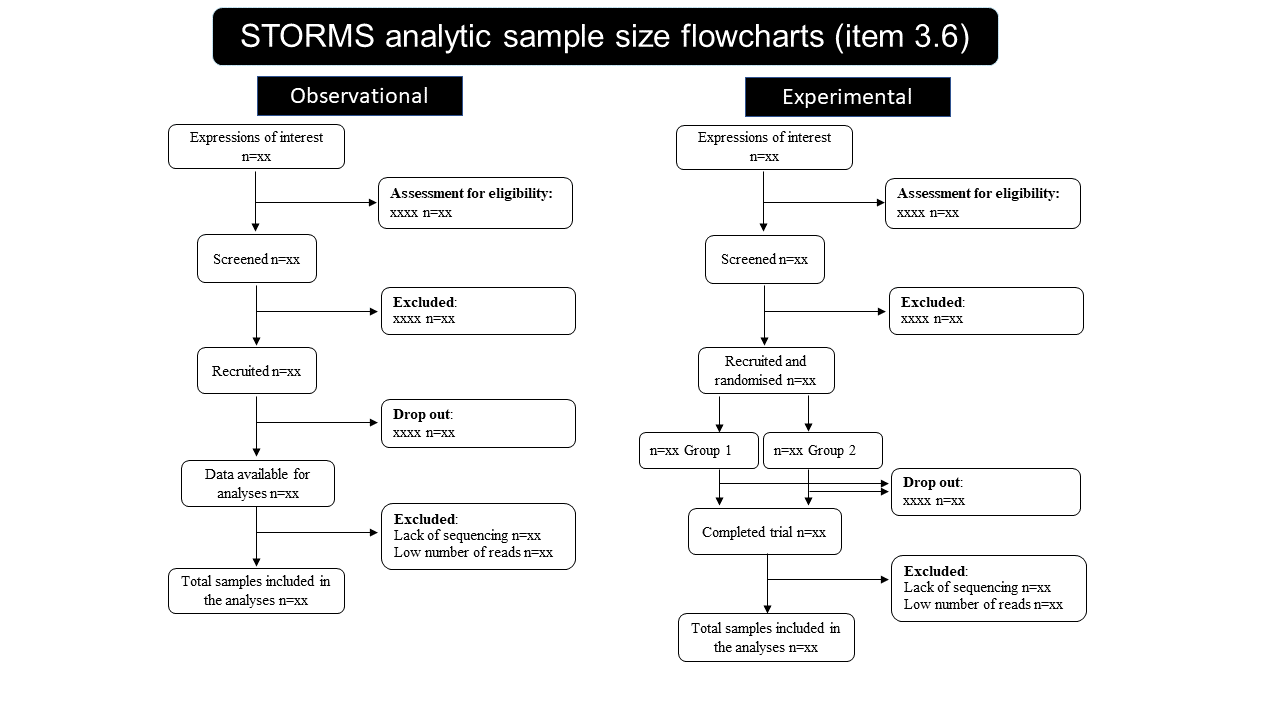
附:
- STROBE指南(Strengthening the Reporting of Observational Studies in Epidemiology)是一个用于规范流行病学观察性研究报告的条目清单。它涵盖了三种主要的研究设计:队列研究、病例对照研究和横断面研究。STROBE指南旨在提供全面和完善的报告标准,以改善研究的方法学问题,并帮助研究者更明智地决定何时开展新研究及其目标。
- 加强遗传关联性研究报告质量 (Strengthening the Reporting of GAS, STREGA)声明即为旨在加强该类研究报告透明化和提高其报告质量的指南。
参考:
- https://streamsmicrobiome.org/
- https://streamsmicrobiome.org/wp-content/uploads/2025/03/streams_simplified.xlsx
- Kelliher, J.M., Mirzayi, C., Bordenstein, S.R. et al. STREAMS guidelines: standards for technical reporting in environmental and host-associated microbiome studies. Nat Microbiol 10, 3059–3068 (2025). https://doi.org/10.1038/s41564-025-02186-2
- https://dmptool.org/plan_from_funder_requirements?org_autocomplete%5Bfunder_id%5D=14176&org_autocomplete%5Bfunder_name%5D=STREAMS+Microbiome&org_autocomplete%5Bid%5D=14176&org_autocomplete%5Bname%5D=STREAMS+Microbiome&plan%5Btemplate_id%5D=2635 (需要注册)
- https://www.stormsmicrobiome.org/
- https://doi.org/10.5281/zenodo.5703116

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐

 22
22 0
0- 0



 已为社区贡献11条内容
已为社区贡献11条内容

所有评论(0)