单细胞转录组分析(具体步骤)
单细胞转录组分析(具体步骤1)
看一下数据是什么样子(一般是公司给的):
10×:
以10X Genomics的单细胞测序数据为例,当公司完成测序后,通常会提供一组原始数据文件。拿到数据后,我们可以在提供的文件夹(如“Data”文件夹)中找到四个示例数据集(如 R8081、R8082、R8083 等)。这些数据集的结构基本相同,主要包含FASTQ格式的原始测序数据。
服务公司通常会使用 Cell Ranger 进行标准化分析,将 FASTQ 文件比对到参考基因组(如 hg38 或 hg19)。在进行数据处理时,需要明确基因组版本,因为这会影响后续的分析和文章撰写。一般来说,hg38 是目前常用的基因组版本。
单细胞数据的关键文件
当 Cell Ranger 处理完成后,输出的结果文件夹中会包含多个文件和子文件夹。其中,最重要的文件位于 filtered_feature_bc_matrix 目录下
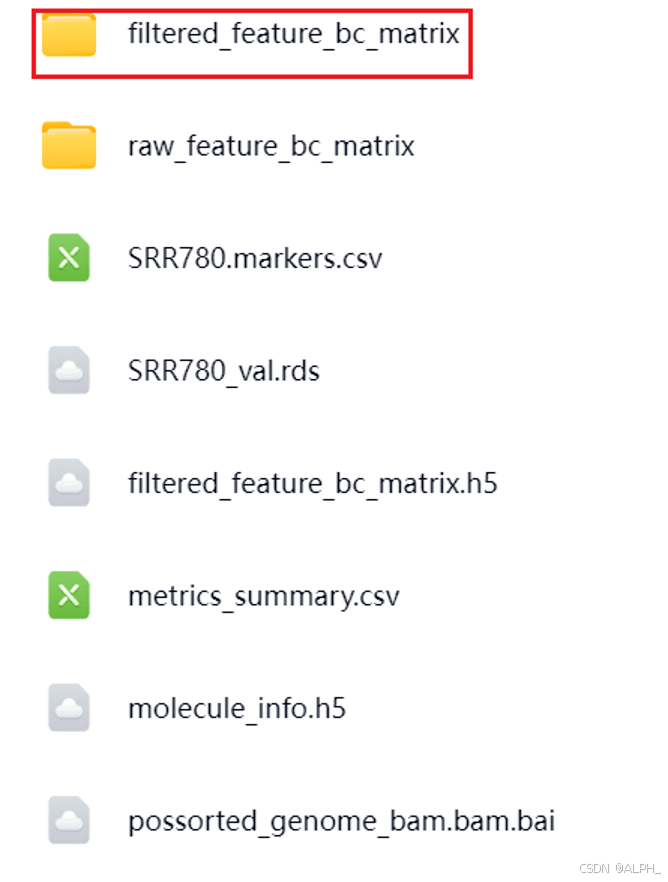
该目录包含三个核心文件:
- barcodes.tsv.gz —— 细胞条形码信息,每一行代表一个单细胞的唯一标识。
- features.tsv.gz —— 基因特征文件,列出了所有被检测到的基因名称。
- matrix.mtx.gz —— 表达矩阵,存储了细胞(barcode)与基因(features)之间的表达量数据,通常是一个稀疏矩阵(Sparse Matrix)。
这些文件共同构成了单细胞测序数据的基础信息,分析时通常只需要这三个文件即可。

BD:
BD(Becton Dickinson)也有自己的一套单细胞测序平台。经过预处理后,BD 会提供一个 RSEC 校正后的 Molecular Profile(分子特征)CSV 文件。


由于该 CSV 文件较大,我们一般不会直接打开查看,但它的基本格式与 10X Genomics 输出的表达矩阵类似:
- 行(row)代表基因
- 列(column)代表细胞
- 矩阵中的数值代表该细胞对该基因的表达水平(UMI count)
单细胞数据的稀疏矩阵特性
无论是 BD 还是 10X Genomics,它们的单细胞 RNA 测序(scRNA-seq)数据都呈现高度稀疏矩阵(Sparse Matrix)的特点。这意味着:
- 大部分值为零:由于 mRNA 转录的随机性以及技术噪声,在单个细胞中,很多基因根本不会被检测到,因此数据矩阵中大多数数值为 0。
- 每个细胞通常检测到几千个基因:尽管整张矩阵可能包含 2~3 万个基因,但每个单细胞通常只会表达其中的几千个。
- 表达量数据为整数(count data):通常是 UMI 计数值(Unique Molecular Identifier Count),如 1、5、10、50 等,而不是连续变量。这是因为 scRNA-seq 通过 UMI 去除 PCR 扩增偏倚,最终测得的是基因的真实拷贝数。
Smart-seq:
Smart-seq 是另一种单细胞 RNA 测序(scRNA-seq)技术,与 10X Genomics 和 BD 不同,它的测序方法更接近于传统的 bulk RNA-seq。
Smart-seq 数据的基本格式
- 细胞命名方式不同:
- 在 10X Genomics 数据中,每个细胞通常使用条形码(Barcode)进行标记。
- 在 Smart-seq 数据中,每个细胞有一个更具体的名称,而不是一个简单的条形码。
- 行(row)代表基因,列(column)代表细胞,与其他 scRNA-seq 数据类似。
- 数据存储格式为 RPKM/TPM/FPKM:
- 由于 Smart-seq 采用的是全长转录组测序(Full-length Transcript Sequencing),而不是 3' 端测序,因此它的基因表达数据通常经过标准化,表现为 RPKM(Reads Per Kilobase per Million Mapped Reads)或 TPM(Transcripts Per Million)。
- 这与 10X Genomics 使用的 UMI 计数(count-based data)不同。
Smart-seq 数据的处理方式
- 在 R 语言环境中,我们可以使用 Seurat 或 Scanpy 进行 Smart-seq 数据分析。
- 由于 Smart-seq 检测到的基因数量较多,因此它的处理方式更接近 bulk RNA-seq,需要额外的标准化步骤。
计算资源需求:
- 处理 Smart-seq 原始 FASTQ 数据需要进行 比对(Alignment) 和 定量(Quantification),通常使用 STAR + RSEM 或 HISAT2 + StringTie。
- 由于计算量较大,通常需要 Linux 服务器 或 高性能计算集群(HPC) 进行数据分析。
- 10X Genomics 提供的 Cell Ranger 只能运行在 Linux 系统上,Windows 和 macOS 用户无法直接运行该流程。
软件安装:
####安装R语言和Rstudio###
#R software: R: The R Project for Statistical Computing
#Rstudio:https://www.rstudio.com
####设置镜像,提高软件包安装速度###
options(BioC_mirror="https://mirrors.tuna.tsinghua.edu.cn/bioconductor")
options("repos" = c(CRAN="https://mirrors.tuna.tsinghua.edu.cn/CRAN/"))
####安装软件包安装的协助包###
###从CRAN安装
install.packages("stringr")###处理字符文件
install.packages("future")##并行计算
install.packages("future.apply")##并行计算
install.packages("ggplot2")###安装作图软件
install.packages("Seurat")###单细胞处理包
install.packages("dplyr")###管道符号
install.packages("ggpubr")###作图与统计
install.packages("shinythemes")###Shiny服务器
install.packages("ggsci")###配色图
library(stringr)
library(future)
library(future.apply)
library(ggplot2)
library(Seurat)
library(dplyr)
library(ggpubr)
####从Bioconductor 安装软件
install.packages("BiocManager")####从Bioconductor网站安装生物信息学软件包
library(BiocManager)
BiocManager::install("limma")
BiocManager::install("clusterProfiler")###进行GO分析
BiocManager::install("ComplexHeatmap")#,force = T)###heatmap作图
BiocManager::install("monocle")##使用此版本monocle2
BiocManager::install("org.Hs.eg.db")
BiocManager::install("infercnv",force = T)
BiocManager::install(c("AUCell", "RcisTarget"))
BiocManager::install(c("GENIE3")) # Optional. Can be replaced by GRNBoost
## Optional (but highly recommended):
# To score the network on cells (i.e. run AUCell):
BiocManager::install(c("zoo", "mixtools", "rbokeh"))
# For various visualizations and perform t-SNEs:
BiocManager::install(c("DT", "NMF", "ComplexHeatmap", "R2HTML", "Rtsne"))
# To support paralell execution (not available in Windows):
BiocManager::install(c("doMC", "doRNG"))
#如果出现Update all/some/none? [a/s/n]: 输入a,回车键
#如果出现Do you want to install from sources the packages which need compilation? (Yes/no/cancel) :输入no,回车键
library(infercnv)
library(limma)
library(clusterProfiler)
library(ComplexHeatmap)
###从Github安装软件
install.packages("devtools")####首先从github网站安装生物信息学软件包##
#library(devtools)
install_github("sqjin/CellChat")
###安装DoubletFinder检测Doublets###
#来自:https://github.com/ddiez/DoubletFinder
devtools::install_github("sqjin/CellChat")
devtools::install_github('chris-mcginnis-ucsf/DoubletFinder')
devtools::install_github("ggjlab/scHCL")
devtools::install_github("immunogenomics/harmony")
devtools::install_github("aertslab/SCopeLoomR", build_vignettes = TRUE)
devtools::install_github("aertslab/SCENIC")
devtools::install_github("sqjin/CellChat")##cell-cell interaction
library(DoubletFinder)
####存在并行计算时###
library(future)
library(future.apply)
plan("multiprocess", workers = 2) ###compute cores计算核心
options(future.globals.maxSize = 8000 * 1024^2)##最大设置内存占用大小
处理10×数据:
####首先认识数据结构####
###10XGenomics 的filtered_feature_bc_matrix
##https://www.ncbi.nlm.n[ih.gov/geo/query/acc.cgi?acc=GSE134520
###BD RSEC_MolsPerCell.csv
###SMART-seq RPKM.txt
rm(list = ls())
#install.packages("Seurat")
####首先我们先处理10X Genomics的数据
library(Seurat)##version 4.0.5
library(dplyr)
library(future)
library(future.apply)
library(DoubletFinder)
plan("multicore", workers = 3) #三个线程
options(future.globals.maxSize = 10000 * 1024^2)#10g内存
#getwd()
setwd("~/Documents_PC/scRNA-seq/Data")
setwd("./10X/")
setwd("~/Documents_PC/scRNA-seq/Data/10X")
list.files(path = "./",pattern = "^SRR")#看一下路径下的文件
####SRR780####
SRR780<-Read10X(data.dir = "SRR80_outs/filtered_feature_bc_matrix/")
SRR780[1:5,1:5]#取前五行和前五列看看
SRR780_object<- CreateSeuratObject(counts = SRR780, project = "SRR780", min.cells = 0, min.features = 200)#一些筛选标记,所有基因导入,一个细胞里必须有200个基因才行
SRR780_object[["percent.mt"]] <- PercentageFeatureSet(SRR780_object, pattern = "^MT-")#所有的线粒体基因都是以MT打头的,看看线粒体基因
hist(SRR780_object[["percent.mt"]]$percent.mt)
VlnPlot(SRR780_object, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)#画一个小提琴图
SRR780_object
plot1 <- FeatureScatter(SRR780_object, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(SRR780_object, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")#画两个图看看质量怎么样,比较高counts和比较高gene数都要去掉
CombinePlots(plots = list(plot1,plot2))
SRR780_object
##select the cells with 300 genes at least and 4000 at most, the percent of mitochondrion genes is less than 10%
SRR780_val<- subset(SRR780_object, subset = nFeature_RNA > 200 & nFeature_RNA < 7000 & percent.mt < 20)#这个就是筛选过程
SRR780_val@meta.data[1:5,]#显示前五行
SRR780_val@meta.data[1:5,]
dim(SRR780_val@meta.data)
colnames(SRR780_val)
rownames(SRR780_val@meta.data)
colname<-paste("SRR780_",colnames(SRR780_val),sep="")
colname
SRR780_val<-RenameCells(object = SRR780_val,colname)
#rm(list = c("BC2","bc2_object"))
SRR780_val@meta.data[1:5,]
SRR780_val$tissue_type<-"Normal"#增加一列
SRR780_val@meta.data[1:5,]
SRR780_val <- NormalizeData(SRR780_val, normalization.method = "LogNormalize", scale.factor = 10000)
SRR780_val <- FindVariableFeatures(SRR780_val, selection.method = "vst", nfeatures = 2000)#找细胞间表达差异大的基因,top2000
SRR780_val@assays$RNA@var.features
length(SRR780_val@assays$RNA@var.features)
###scaling the data###
all.genes <- rownames(SRR780_val)
all.genes[1000:10010]
SRR780_val <- ScaleData(SRR780_val,vars.to.regress = c("percent.mt"))
#去除线粒体基因的一个占比,剩下的基因进行归一化
SRR780_val
###perform linear dimensional reduction###
SRR780_val <- RunPCA(SRR780_val, features = VariableFeatures(object = SRR780_val))#用PCA降维
print(SRR780_val[["pca"]], dims = 1:5, nfeatures = 5)
#dev.off()
VizDimLoadings(SRR780_val, dims = 1:2, reduction = "pca")
ElbowPlot(SRR780_val,ndims = 50)
####cluster the cells###
SRR780_val <- FindNeighbors(SRR780_val, dims = 1:30)#取到20或者拐点之后,区别不大,在30维度区间上计算哪些细胞之间是更相似的,聚集在一块
####Run non-linear dimensional reduction (UMAP/tSNE)###
SRR780_val <- RunUMAP(SRR780_val, dims = 1:30)
SRR780_val<-RunTSNE(SRR780_val,dims=1:30)
SRR780_val <- FindClusters(SRR780_val, resolution = 1)#这块是1,根据基因表达来定
DimPlot(SRR780_val, reduction = "umap",label = T)
#画一个细胞分群图,靠的近在功能和基因表达上会相近
DimPlot(SRR780_val, reduction = "tsne",label = T)
table(Idents(SRR780_val))
getwd()
SRR780.markers <- FindAllMarkers(SRR780_val, only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)#找高表达差异基因,在细胞里面比例25%
write.csv(SRR780.markers,"./SRR80_outs/SRR780.markers.csv")
marker <- FindMarkers(SRR780_val, ident.1 = "3",ident.2 = "7",only.pos = TRUE, min.pct = 0.25, logfc.threshold = 0.25)
######biomarkers###
###epithelial cell
FeaturePlot(SRR780_val,features = c("EPCAM","CDH1","KRT7","KRT5","UPK1A","KRT20"),reduction = "tsne",label = T)
##TNK cell
FeaturePlot(SRR780_val,features = c("CD3D","CD8A","CD4","NCAM1"),reduction = "tsne",label = T)
##endothelial cell
FeaturePlot(SRR780_val,features = c("VWF","PECAM1"),reduction = "tsne",label = T)
####fibroblasts and pericytes
FeaturePlot(SRR780_val,features = c("RGS5","COL1A1","PDGFRA","PDGFRB","DES"),reduction = "tsne",label = T)
###B cell and plasma
FeaturePlot(SRR780_val,features = c("CD19","JCHAIN","CD79A"),reduction = "tsne",label = T)
###myeloid cell
FeaturePlot(SRR780_val,features = c("CD74","CD14","FCGR3A","S100A8"),reduction = "tsne",label = T)
###mast cell
FeaturePlot(SRR780_val,features = c("CPA3"),reduction = "tsne",label = T)
不同类型的细胞及其标志基因
|
细胞类型 |
标志基因 |
|
上皮细胞(epithelial cell) |
EPCAM, CDH1, KRT7, KRT5, UPK1A, KRT20 |
|
T/NK细胞(TNK cell) |
CD3D, CD8A, CD4, NCAM1 |
|
内皮细胞(endothelial cell) |
VWF, PECAM1 |
|
成纤维细胞及周细胞(fibroblasts and pericytes) |
RGS5, COL1A1, PDGFRA, PDGFRB, DES |
|
B细胞和浆细胞(B cell and plasma cell) |
CD19, JCHAIN, CD79A |
|
髓系细胞(myeloid cell) |
CD74, CD14, FCGR3A, S100A8 |
|
肥大细胞(mast cell) |
CPA3 |
SRR780_val<-RenameIdents(SRR780_val,"3"="Epithelial","10"="Epithelial","1"="Fibroblast","4"="Pericytes","9"="Pericytes","13"="MAST","5"="B",
"11"="Plasma_B","6"="T/NK","2"="T/NK","8"="Myeloid","0"="Endothelial","12"="Endothelial","7"="Unknown")
DimPlot(SRR780_val,reduction = "tsne",label = FALSE)
DimPlot(SRR780_val,reduction = "umap",label = T)
table(Idents(SRR780_val))###查看有多少个细胞
SRR780_val@meta.data[1:5,]
table(Idents(SRR780_val))
Idents(SRR780_val)<-SRR780_val$RNA_snn_res.0.6
table(Idents(SRR780_val))
SRR780_val$cell_cluster_origin<-Idents(SRR780_val)
SRR780_val@meta.data[1:5,]
saveRDS(SRR780_val,file = "./SRR80_outs/SRR780_val.rds")
#save.image(file="./SRR780_outs/SRR780_val.RData")
#load("./SRR780_outs/SRR780_val.RData")
SRR780_val$cluster<-Idents(SRR780_val)
table(Idents(SRR780_val))
####auto cellular annotation#####
#install.packages("shinythemes")
#devtools::install_github("ggjlab/scHCL")###郭国冀老师团队
library(scHCL)#自动注释软件包
hcl_result <- scHCL(scdata = SRR780_val@assays$RNA@data, numbers_plot = 1)###time comsuming take several mins
hcl_result$scHCL#预测结果
cellassign<-table(Idents(SRR780_val),hcl_result$scHCL_probility$`Cell type`)
cellassign
cellassign<-table(Idents(SRR780_val),hcl_result$scHCL)
cellassign
#SRR780_val$RNA_snn_res.0.6
cellassign<-table(SRR780_val$RNA_snn_res.0.6,hcl_result$scHCL_probility$`Cell type`)
cellassign
library(stringr)
cell_predict<-data.frame(hcl_result$scHCL_probility)
SRR780_val@meta.data[1:5,]
table(rownames(SRR780_val@meta.data)==cell_predict$Cell)
str_split(cell_predict$Cell.type,"[.]",simplify = T)[,1]
cell_predict$cell_predict<-str_split(cell_predict$Cell.type,"[.]",simplify = T)[,1]
table(SRR780_val$RNA_snn_res.0.6,cell_predict$cell_predict)
SRR780_val$cell_predict<-cell_predict$cell_predict
SRR780_val@meta.data[1:3,]
table(Idents(SRR780_val))
SRR780_val$cluster<-Idents(SRR780_val)
####用singleR软件来注释细胞#### 有空闲时跑
#devtools::install_github("dviraran/SingleR")
BiocManager::install("SingleR")
BiocManager::install("celldex")
library(SingleR)
library(celldex)
sc_data<-as.matrix(SRR780_val@assays$RNA@counts)
hpca.se <- HumanPrimaryCellAtlasData()
hpca.se$label.main
pred.hesc <- SingleR(test = sc_data, ref = hpca.se, assay.type.test=1,
labels = hpca.se$label.main)
pred.hesc$labels###预测的结果
####Doublets identification####
library(DoubletFinder)
###完整的分析:前面和建立单细胞Seurat对象一样,因为前面跑过了,这里都省略
if (TRUE){
SRR780<-Read10X(data.dir = "SRR80_outs//filtered_feature_bc_matrix/")
SRR780[1:5,1:5]
SRR780_object<- CreateSeuratObject(counts = SRR780, project = "SRR780", min.cells = 0, min.features = 200)
SRR780_object[["percent.mt"]] <- PercentageFeatureSet(SRR780_object, pattern = "^MT-")
hist(SRR780_object[["percent.mt"]]$percent.mt)
VlnPlot(SRR780_object, features = c("nFeature_RNA", "nCount_RNA", "percent.mt"), ncol = 3)
plot1 <- FeatureScatter(SRR780_object, feature1 = "nCount_RNA", feature2 = "percent.mt")
plot2 <- FeatureScatter(SRR780_object, feature1 = "nCount_RNA", feature2 = "nFeature_RNA")
CombinePlots(plots = list(plot1,plot2))
SRR780_object
##select the cells with 300 genes at least and 4000 at most, the percent of mitochondrion genes is less than 10%
SRR780_val<- subset(SRR780_object, subset = nFeature_RNA > 200 & nFeature_RNA < 7000 & percent.mt < 20)
SRR780_val@meta.data[1:5,]
colname<-paste("SRR780_",colnames(SRR780_val),sep="")
colname
SRR780_val<-RenameCells(object = SRR780_val,colname)
#rm(list = c("BC2","bc2_object"))
SRR780_val@meta.data[1:5,]
SRR780_val$tissue_type<-"Normal"
SRR780_val <- NormalizeData(SRR780_val, normalization.method = "LogNormalize", scale.factor = 10000)
SRR780_val <- FindVariableFeatures(SRR780_val, selection.method = "vst", nfeatures = 2000)
###scaling the data###
all.genes <- rownames(SRR780_val)
SRR780_val <- ScaleData(SRR780_val,vars.to.regress = c("percent.mt"))
SRR780_val
###perform linear dimensional reduction###
SRR780_val <- RunPCA(SRR780_val, features = VariableFeatures(object = SRR780_val))
print(SRR780_val[["pca"]], dims = 1:5, nfeatures = 5)
#dev.off()
VizDimLoadings(SRR780_val, dims = 1:2, reduction = "pca")
ElbowPlot(SRR780_val,ndims = 50)
####cluster the cells###
SRR780_val <- FindNeighbors(SRR780_val, dims = 1:30)
####Run non-linear dimensional reduction (UMAP/tSNE)###
SRR780_val <- RunUMAP(SRR780_val, dims = 1:30)
SRR780_val<-RunTSNE(SRR780_val,dims=1:30)
SRR780_val <- FindClusters(SRR780_val, resolution = 0.6)
}
SRR780_val@meta.data[1:5,]
####因为前面跑过,所以直接从这步开始计算###
saveRDS(SRR780_val,file="SRR780.rds")
SRR780_val_db<-SRR780_val
sweep.res.list_SRR780_val <- paramSweep_v3(SRR780_val_db, PCs = 1:30)
sweep.stats_SRR780_val <- summarizeSweep(sweep.res.list_SRR780_val, GT = FALSE)
bcmvn_SRR780_val<-find.pK(sweep.stats_SRR780_val)
pK_value <- as.numeric(as.character(bcmvn_SRR780_val$pK[bcmvn_SRR780_val$BCmetric == max(bcmvn_SRR780_val$BCmetric)]))
annotations <- SRR780_val_db@meta.data$RNA_snn_res.0.6
homotypic.prop <- modelHomotypic(annotations) ####get the estimated number
nExp_poi <- round(homotypic.prop*length(SRR780_val_db@meta.data$orig.ident))
nExp_poi.adj <- round(nExp_poi*(1-homotypic.prop))
pN_value <- 0.25
pANN_value <- paste0("pANN_",pN_value,"_",pK_value,'_',nExp_poi)
SRR780_val_db <- doubletFinder_v3(SRR780_val_db, PCs = 1:30, pN = pN_value, pK = pK_value, nExp = nExp_poi, reuse.pANN = FALSE)
SRR780_val_db <- doubletFinder(SRR780_val_db, PCs = 1:30, pN = pN_value, pK = pK_value, nExp = nExp_poi.adj, reuse.pANN = pANN_value)
SRR780_val_db@meta.data[1:5,]
SRR780_val$doublets<-SRR780_val_db$DF.classifications_0.25_0.3_83
SRR780_val@meta.data[1:5,]
####saveRDS###
saveRDS(SRR780_val,file = "./SRR80_outs/SRR780_val.rds")
DimPlot(SRR780_val,reduction = "tsne",group.by = "doublets")
DimPlot(SRR780_val,reduction = "tsne",group.by = "cluster",label = T)
DimPlot(SRR780_val,reduction = "tsne",label = T)

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐

 43
43 0
0- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)