ChAMP 差异甲基化分析
上一篇写了用champ.load导入甲基化数据,成功导入之后就可以做一系列的流程了,我主要是根据ChAMP包的说明和生信技能树的甲基化芯片分析来学习这个整体流程的。首先是安装包# 首先要有Bioconductor ,然后用代码:If(!requireNamwspace(“BiocManager”,quietly=TRUE))Install.packages(“BiocManager”)BiocMa
上一篇写了用champ.load导入甲基化数据,成功导入之后就可以做一系列的流程了,我主要是根据ChAMP包的说明和生信技能树的甲基化芯片分析来学习这个整体流程的。
首先是安装包
# 首先要有Bioconductor ,然后用代码:
If(!requireNamwspace(“BiocManager”,quietly=TRUE))
Install.packages(“BiocManager”)
BiocManager::install(“ChAMP”)
# 安装后就可以加载ChAMP包
library("ChAMP")
这个包里含有测试数据集,450k 肺肿瘤数据集,仅包含 8 个样本,4 个肺肿瘤样本 T 和 4 个对照样本 C,这可以通过将目录指向 testDataSet 来加载,如下:
testDir=system.file("extdata",package="ChAMPdata")
myLoad<-champ.load(testDir,arraytype= "450k")
# 使用可以使用一个命令一次运行完整的管道:
champ.process(directory = testDir)
# 分部的运行
myLoad <- cham.load(testDir) # 导入
# 这一步还可以分为 champ.import(testDir) + champ.filter() 过滤探针用的
CpG.GUI()
champ.QC() # Alternatively: QC.GUI() 质控
myNorm <- champ.norm() # 归一化,得到beta值矩阵
champ.SVD() # 奇异值分解
# 如果发现有批次效应, 可以用 champ.runCombat() 消除.
myDMP <- champ.DMP() # 差异位点
DMP.GUI()
myDMR <- champ.DMR() # 差异区域
DMR.GUI()
myBlock <- champ.Block() # 差异块
Block.GUI()
myGSEA <- champ.GSEA() # 富集分析
myCNA <- champ.CNA() # 拷贝数变异
对测试数据做完一套流程,结合对数据的了解,大致能明白每一步是干嘛的,下面根据ChAMP手册理一下每一个部分。
1.导入数据
这一步上一篇有比较详细的说明过,那个Sample_Sheet.csv文件导入后会被命名为pd文件,即表型文件,其中的样本类型(Sample_Group或者是Sample_Type)是其中比较重要的一个信息
myLoad$pd
测试数据中Sample_Group 包含两种表型:C 和 T, C 表示“对照”,而 T 表示“肿瘤”。
这一步中还同时做了Filtering 即过滤数据,这个函数可以过滤掉一些不合格的探针,ChAMP手册中有六个过滤的条件,450k初始有48万多探针,这一步之后还有41万多了。我在做的时候没有单独去做Filtering ,直接用cham.load导入了。
2.质量控制
质量控制是确保数据集适合下游分析的重要过程。 champ.QC() 函数和 QC.GUI() 函数会为用户绘制一些图,以方便检查他们的数据质量。 通常,会生成三个图:
champ.QC()
mdsPlot(多维缩放图):该图允许基于所有样本中前 1000 个最可变探针的样本相似性可视化。 在此图中,样本按样本组着色。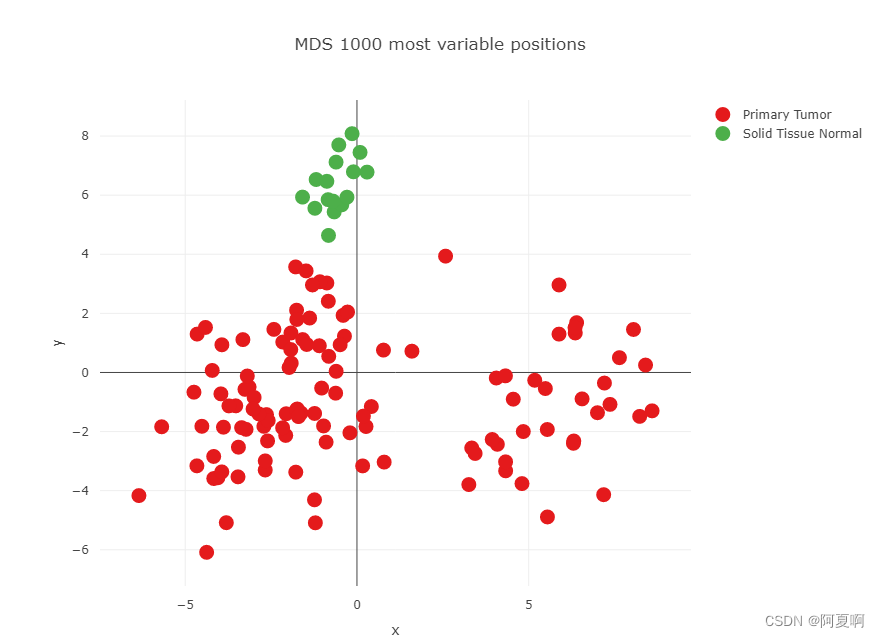
可以看出来Tumor和Normal还是分得开的
densityPlot:每个样本的 beta 分布; 用户可以使用该图查找与其他样本有显着差异且质量可能不佳(例如亚硫酸氢盐转化不完全)的样本。 注意:它还用于识别和确认甲基化或未甲基化的对照样品(如果这些样品包括在研究中)。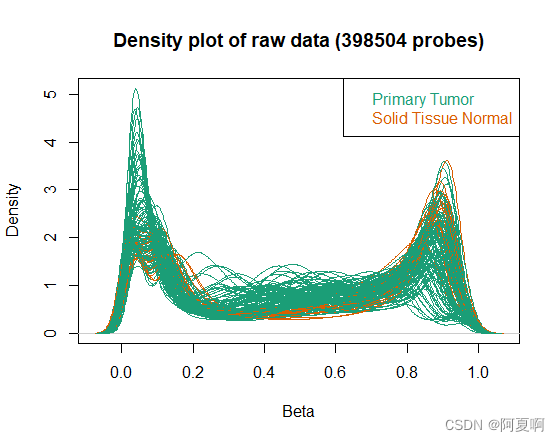
dendrogram:所有样本的聚类图,您可以选择不同的方法来生成此图。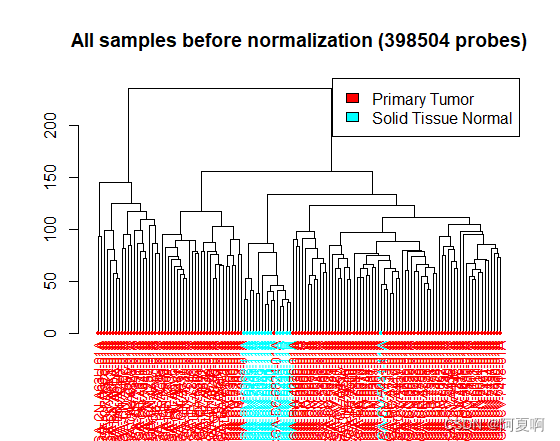
QC.GUI(beta=myLoad$beta,arraytype="450K")
与 champ.QC() 相比,QC.GUI() 将提供五个交互式绘图。 这些图如下:mdsPlot、type-I&II densityPlot、样本 beta 分布图、树状图和前 1000 个信息最多的 CpG 热图。 可以对这些图进行交互,以查看样品是否符合进一步分析的条件,并检查聚类结果。
Probe-Type-I&II plot:数据集中 Type-I 和 Type-II 探针的 beta 分布,可以检查数据集的标准化状态。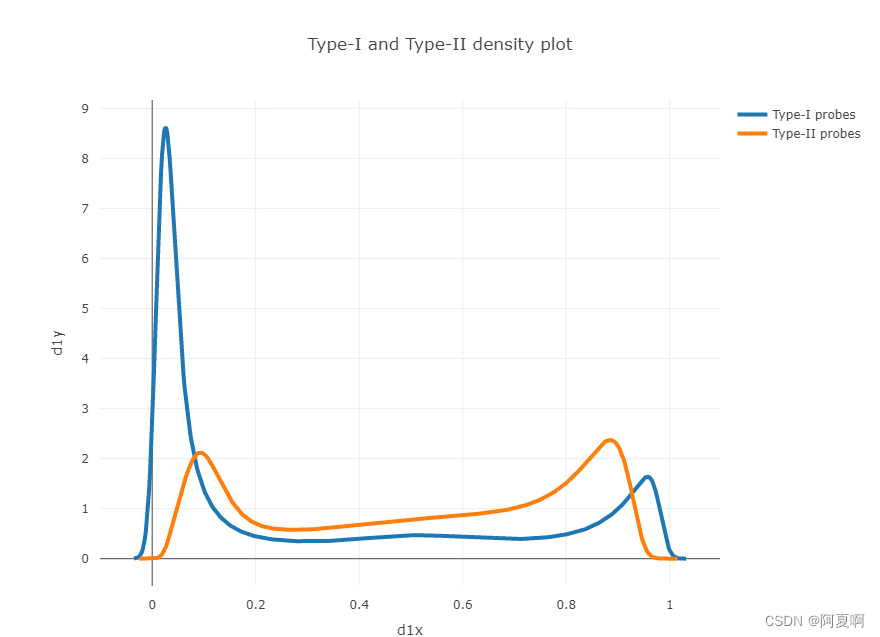
顶部变量 CpG 的热图:此热图将选择信息含量最多的 CpG,并根据这些位点的甲基化值创建热图。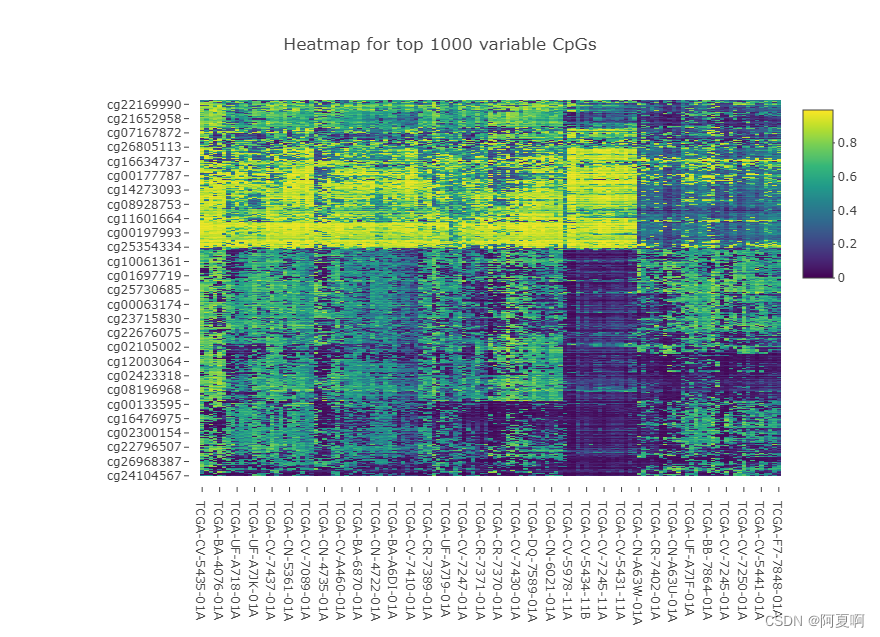
这个热图在测试数据中表现很好,但是在我的数据中表现不好,可能是因为我的Tumor和Normal数量差的过多,上面的图横轴中11结尾的是Tumor,01结尾的是Normal,也可能还有别不太清楚的原因,后续在慢慢学。
3.归一化
myNorm <- champ.norm(beta=myLoad$beta,arraytype="450K",cores=5)
输入beta值,阵列类型,如果计算机有多个内核,可以在函数中设置参数“cores”以加速函数运行速度。然后就可以算出归一化后的beta值了。
标准化后,可以使用 QC.GUI() 函数再次检查结果; 记得将 QC.GUI() 函数中的 beta 参数设置为“myNorm”。结果还有有区别的。
很多时候是可以直接下载到标准的beta值矩阵的,生信技能树讲的一个案例就是直接用beta值做的,其中也给到了几个做质控的方法,一个PCA的聚类图,还有两个热图(应该是聚类和相关性的热图)。
PCA也是明显的分了两类: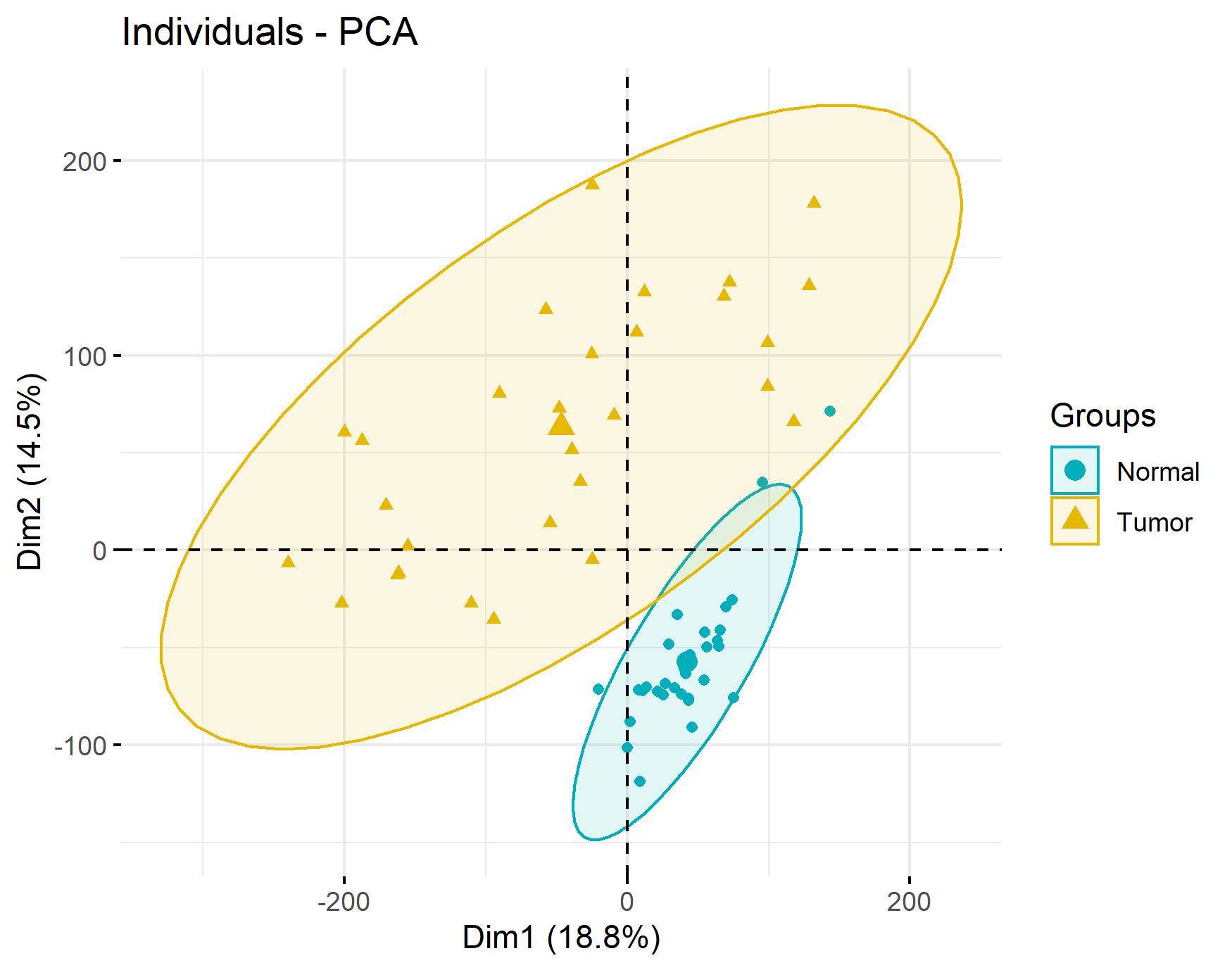
这个应该是根据前一千个探针做的聚类: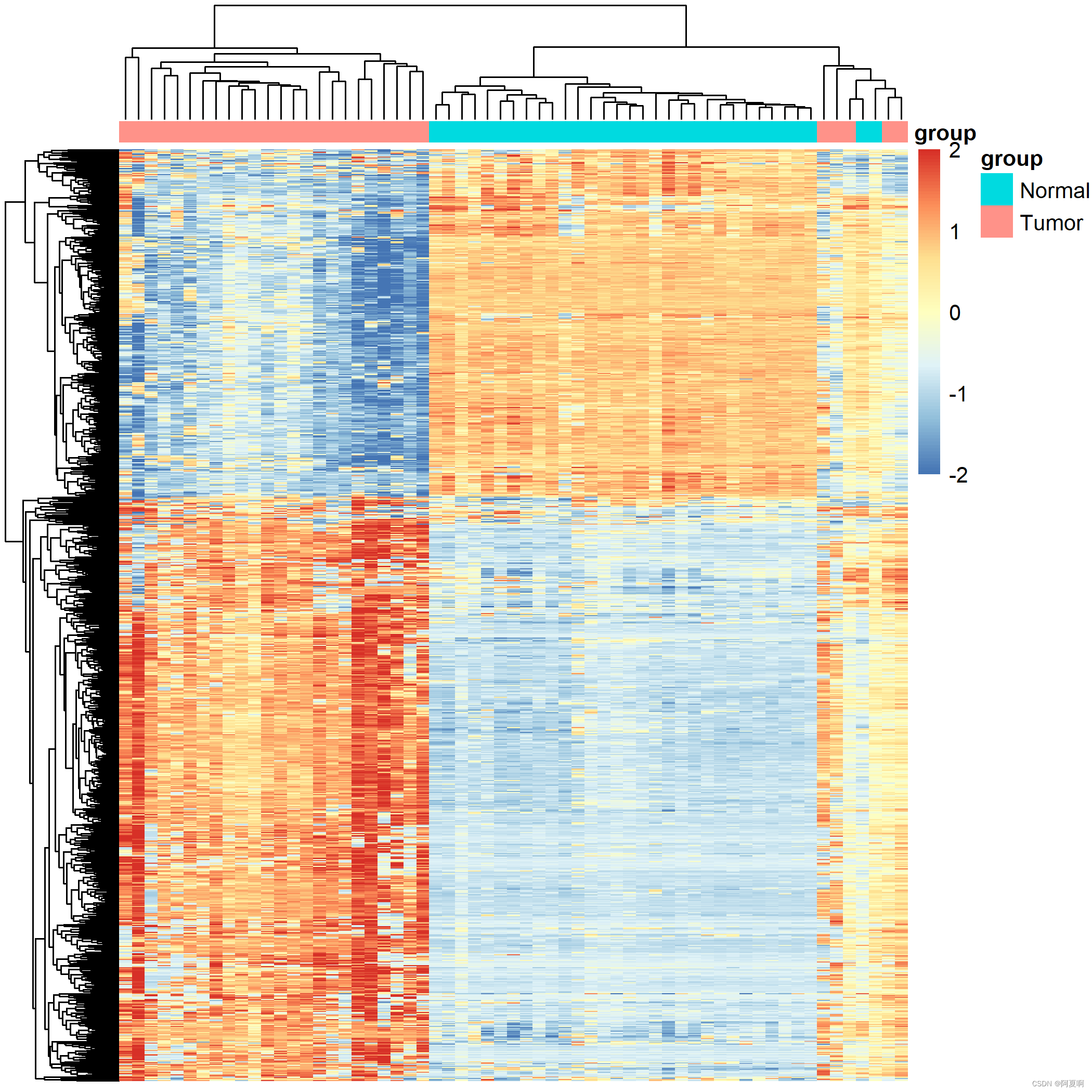
这个是根据前五百个探针做了相关性: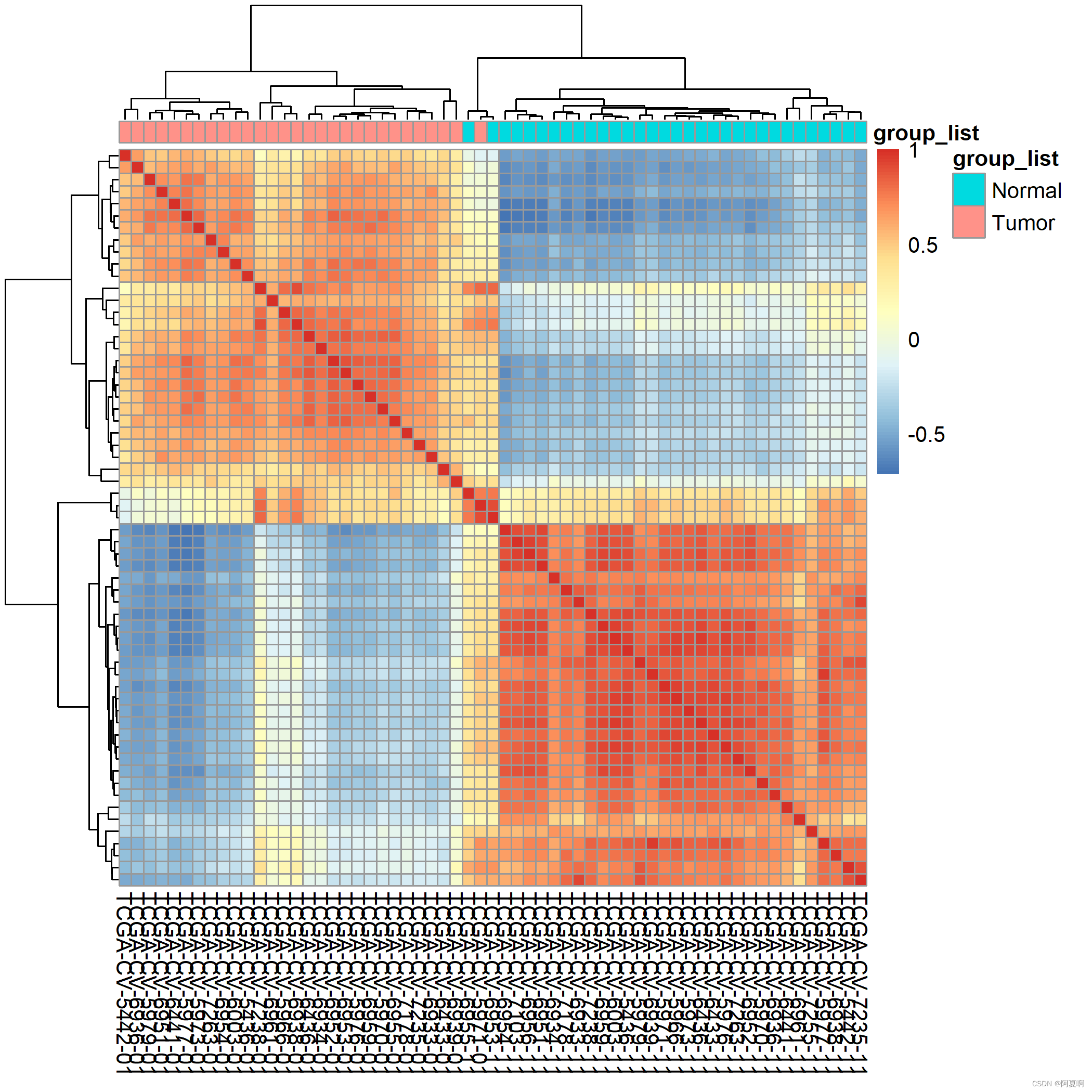
这几个图看起来好像高级一点,做法在B站有视频,代码也是公开的。
4.奇异值分解(SVD)和矫正批次效应
champ.SVD(beta=myNorm,pd=myLoad$pd)
这一部分不是太理解,感觉是观察前二十个主成分和pd文件中协变量之间的相关性,pd中的协变量可以有不同的批次,样本的一些属性信息(流行病学信息),在构造Sample_Sheet.csv的时候是可以加入一些属性信息的。
如果有批次效应的话可以用ComBat 来消除批次效应,需要在函数中设置参数 batchname=c(“Slide”) ,champ.runCombat() 将从批次名称中提取批次,这就需要理解数据的命名方式了,显然我还不太理解,但是也可以先试试
myCombat <- champ.runCombat(beta=myNorm,pd=myLoad$pd,batchname=c("Slide"))
下面是做矫正批次效应之前的SVD: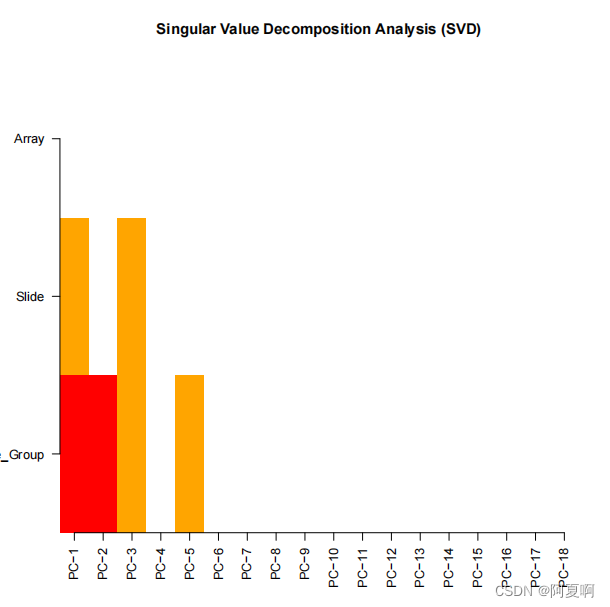
这个是做矫正批次效应之后的SVD: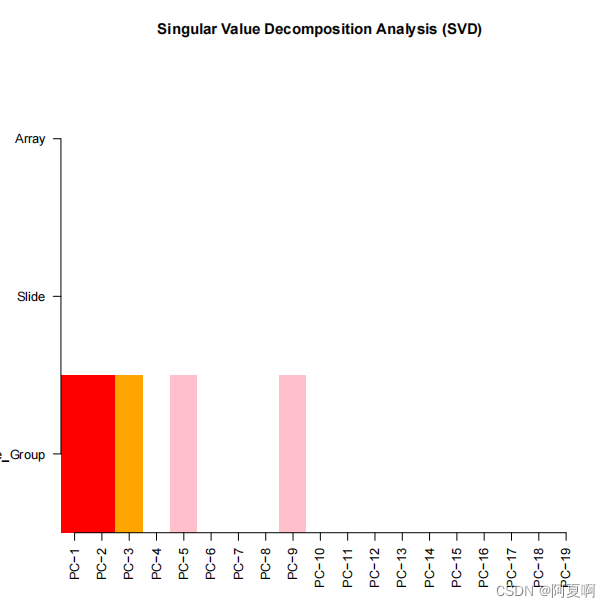
这个图横轴是前二十个主成分,纵轴是pd中的协变量,其中的Slide是我们构造的Sample_Sheet.csv中的Sentrix_ID这一列,也就是批次名。校正之前主成分和批次还是有相关性的,矫正之后就没有了。
5.差异甲基化位点
myDMP <- champ.DMP(beta = myNorm,pheno=myLoad$pd$Sample_Group)
这一部分输入的是beta值和表型文件(就是把样本分成Tumor和Normal的那一列),根据不同的表型求差异。
这一部分比较重要的应该是数据的整理,需要配对的Tumor和Normal样本才可以做,这个可以去看生信技能树的视频,里面还有一些有意思的图,可以根据自己的需求去借鉴他的代码。
最终会得到差异的P值,调整后的P值(FDR),t统计量,logFC,delta beta值等,对后续的筛选有很大的用处。这里的值是什么意思可以去看这个函数的说明书。
之后我想根据差异继续再去选区别不同表型的,最重要的探针,然后做后续的分析,比如通过LASSO或者一些机器学习算法去做筛选,所以做到这一步就停下了,后面的差异区域和差异块没有自己做,GSEA做富集分析的话面临一个网速不够的问题,拷贝数也没有做,如果后面做了的话记得过来记录一下。

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐


 4
4 1
1- 0



 已为社区贡献2条内容
已为社区贡献2条内容

所有评论(0)