【论文阅读】Single-cell assignment using multiple-adversarial domain adaptation network with large-scale r
SELINA是一个基于大规模单细胞转录组参考图谱的自动化细胞类型注释框架。该研究整合了136个数据集、170万细胞和230种人类细胞类型,构建了全面统一的参考图谱。SELINA创新性地采用多重对抗域自适应网络消除批次效应,结合合成少数类过采样技术提高稀有细胞检测能力,并通过自编码器实现查询数据与参考数据的精准匹配。在17个组织95个数据集上的验证表明,SELINA在注释准确性上显著优于现有工具,尤

代码地址:https://github.com/SELINA-team/
摘要
单细胞RNA测序(scRNA-seq)数据的快速积累为描绘多种人类细胞群体提供了丰富资源。然而,利用公共参考数据实现准确的细胞类型注释仍然面临挑战,主要包括注释不一致、批次效应以及稀有细胞类型的存在。为此,我们提出了 SELINA(single-cell identity navigator),这是一个基于预先整理的跨多种组织的参考图谱的综合性、自动化细胞类型注释框架。SELINA 采用多重对抗域自适应网络来消除参考数据集中的批次效应;此外,它通过合成少数类过采样增强了对低频细胞类型的注释能力,并利用自编码器将查询数据与参考数据进行匹配。最终,SELINA 构建了一个覆盖 170 万细胞、涵盖 230 种不同人类细胞类型的全面且统一的参考图谱。我们验证了 SELINA 在多种人类组织中的稳健性和优越性,尤其值得注意的是,它能够在多种疾病背景下实现细胞的准确注释。SELINA 同时提供了 Python 与 R 软件包,为人类单细胞RNA测序数据的注释提供了一套完整的解决方案。
动机
细胞类型注释是解释单细胞转录组测序(scRNA-seq)数据中细胞功能的关键步骤。细胞类型注释主要有两类方法:基于标记基因的方法和基于参考数据的方法。基于参考的方法通过机器学习技术将参考数据集中的细胞类型标签转移到查询数据集中,从而提升了准确性并扩大了应用范围。然而,该领域仍存在一些挑战,包括难以充分利用大规模公共数据、细胞数量不平衡、批次效应以及对参考数据质量的依赖。解决这些挑战对于提高细胞类型注释的准确性、并充分释放scRNA-seq数据的潜力至关重要。
引言
单细胞RNA测序(scRNA-seq)能够对数千个细胞进行分析,从而揭示复杂组织中的异质性。在scRNA-seq数据处理中,细胞类型注释是关键步骤,它对于解释特定细胞类型的功能特征至关重要,并且是许多下游分析(包括轨迹分析和细胞—细胞互作分析)的必要前提。
细胞类型注释方法大致可分为两类:基于标记基因的方法和基于参考数据的方法。前者如 Garnett¹ 和 SCINA²,依赖于聚类表现以及细胞类型特异性标记基因的质量。而后者则通过各种机器学习技术,将参考数据集中的细胞类型标签转移到查询数据集中。代表性方法包括 scmap³、scPred⁴、SingleR⁵、CHETAH⁶、SingleCellNet⁷、ACTINN⁸、mtSC⁹、Cell BLAST¹⁰、CellO¹¹、scCATCH¹²、scMatch¹³、scDeepSort¹⁴ 和 CellTypist¹⁵。由于基于参考的方法不需要先验知识¹⁶,随着scRNA-seq数据的不断积累和通量的提升,它们相比基于标记基因的方法展现出更高的准确性和更广泛的应用价值。
尽管基于参考的方法在细胞类型注释中具有上述优势,但仍存在若干亟需解决的挑战:
-
参考数据利用不足:现有工具多用于单一参考数据与单一查询数据之间的注释迁移,难以充分利用庞大公共数据中蕴含的丰富信息。
-
细胞数量不均衡:不同细胞类型的数量常常不平衡,少数类细胞在建模过程中往往被忽视。
-
批次效应干扰:参考数据与查询数据之间潜在的批次效应往往被忽略,从而影响标签迁移的准确性。
-
过度依赖参考数据质量与覆盖度:例如,ScCATCH 依赖于预训练的细胞类型特异性标记基因;SingleR、CellO 和 scMatch 提供的是来自整体RNA测序(bulk RNA-seq)的参考;而 scDeepSort 和 CellTypist 仅提供有限谱系(如免疫细胞或胚胎细胞)的scRNA-seq参考。
尽管已经有大量努力用于系统收集和整理公共数据集,建立了涵盖数百万细胞的scRNA-seq数据门户,例如 人类细胞图谱(HCA)¹⁷、动物细胞图谱(ACA)¹⁸、Broad研究所的Single Cell Portal¹⁹、人类细胞景观(HCL)²⁰,以及欧洲生物信息研究所(EMBL-EBI)的Single Cell Expression Atlas²¹,但由于注释不一致以及跨数据集存在显著批次效应,目前仍缺乏一个统一且全面的参考图谱。
为了解决这些挑战,我们构建了一个全面的单细胞转录组数据图谱,涵盖来自7种不同测序平台的35个人体组织,共计136个数据集。这些数据集已通过精细整理,并在 HUSCH²² 网站上对外开放。基于 1,706,710个统一处理的细胞(覆盖230种细胞类型),我们提出了一种能够有效利用多数据集进行单细胞注释的算法:
-
采用 合成少数类过采样技术(SMOTE)²³ 增加稀有细胞类型的数量;
-
利用 多对抗域自适应(MADA)²⁴ 在预训练阶段动态更新深度监督学习框架的参数;
-
并通过 自编码器 在预测阶段根据查询数据分布自适应地调整预训练参数。
我们验证了 SELINA(single-cell identity navigator) 在消除批次效应方面的强大能力,并在来自17个组织的95个数据集上系统性评估了其与现有工具的对比性能。此外,我们证明了SELINA能够通过测试 Allen Institute²⁵–²⁷ 的数据集,兼容并利用其他统一数据库作为参考。最后,多种疾病场景下的基准实验显示,SELINA在细胞类型注释的准确性上显著优于主流方法。
凭借其全面的细胞类型参考图谱和卓越的注释迁移能力,SELINA为用户准确进行单细胞注释提供了坚实保障。
方法
SELINA 工作流程主要由两个步骤构成:参考构建和细胞类型预测。
在 参考构建 阶段,我们从多个数据库中收集了公共 scRNA-seq 数据集,并使用 MAESTRO²⁸ 的标准化流程进行处理。该流程包括质量控制、主成分分析(PCA)、数据集内的批次效应去除、无监督聚类,以及基于原始研究中的标签或细胞类型特异性基因标记的注释(图 S1A)。随后,跨数据集的不一致注释被人工统一,并依据 Cell Ontology²⁹ 及相关文献将其划分为主干谱系和次级谱系(图 S1B)。在注释统一后,同一组织内的所有数据集被合并,并利用 Harmony³⁰ 去除了跨数据集的批次效应。针对每个谱系,进一步剔除了异常细胞,以校正潜在的错误注释。最后,经过整理的细胞类型被组织成一个 细胞类型本体树。
在统一的参考基础上,我们开发了一个由三步组成的 细胞类型预测算法:细胞类型平衡、预训练和微调。通常情况下,分类器在训练时可能会以牺牲少数类细胞的准确性为代价来获得更高的整体训练精度。为此,SELINA 利用 SMOTE²³ 生成合成样本,以提高少数类细胞的权重,从而增强分类器对少数类细胞的敏感性。随后,SELINA 以某一组织的数据集作为输入,采用基于 MADA²⁴ 的网络来获取预训练模型。通过在对抗方式下训练深度监督学习框架,能够揭示来自不同测序平台但属于相同细胞的潜在共性信息。为了进一步去除参考数据与查询数据之间的批次噪声,SELINA 使用 自编码器 根据查询数据的分布自适应地微调预训练参数。最终,参考数据集中的标签将通过完整训练的模型准确转移到查询数据中(图 1)。
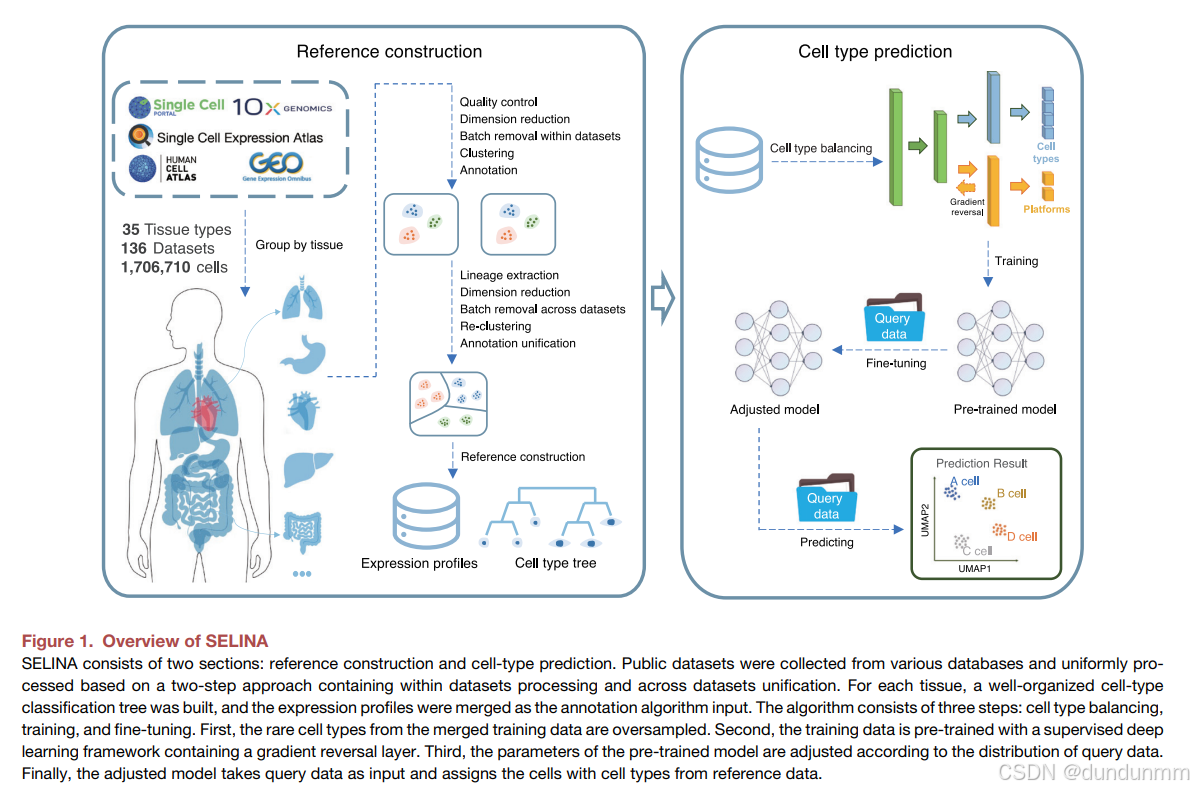
SELINA 提供了大规模、精确注释的人类单细胞表达参考
我们共收集了 1,706,710 个细胞,来源于 136 个数据集,用于构建单细胞转录组数据门户,覆盖 230 种人类细胞类型 和 7 种不同测序平台(表 S1)。在 人类细胞景观(HCL)²⁰ 的基础上,我们进一步扩展了参考图谱,纳入了来自其他测序平台的更多数据集。所有数据集被划分为 35 种主要组织,其定义与 HCL 保持一致。
我们首先总结了参考数据集的特征:血液(n = 14)、肠道(n = 14)和骨髓(n = 13)拥有最多的数据集(图 2A)。血液和骨髓组织还包含了最多的细胞(图 S2B),这表明我们的参考对免疫细胞具有更好的刻画能力;肾脏则具有最丰富的细胞类型(图 S2A)。值得注意的是,35 种组织中有 27 种组织在参考中包含了两个或以上数据集,这表明参考图谱具有较好的覆盖度和深度。大多数数据由 10x Genomics 和 Microwell-seq 生成,分别包含 60 个和 58 个数据集;而 Smart-seq、InDrop、Drop-seq 和 snDrop-seq 的数据仅占少部分(图 2A)。
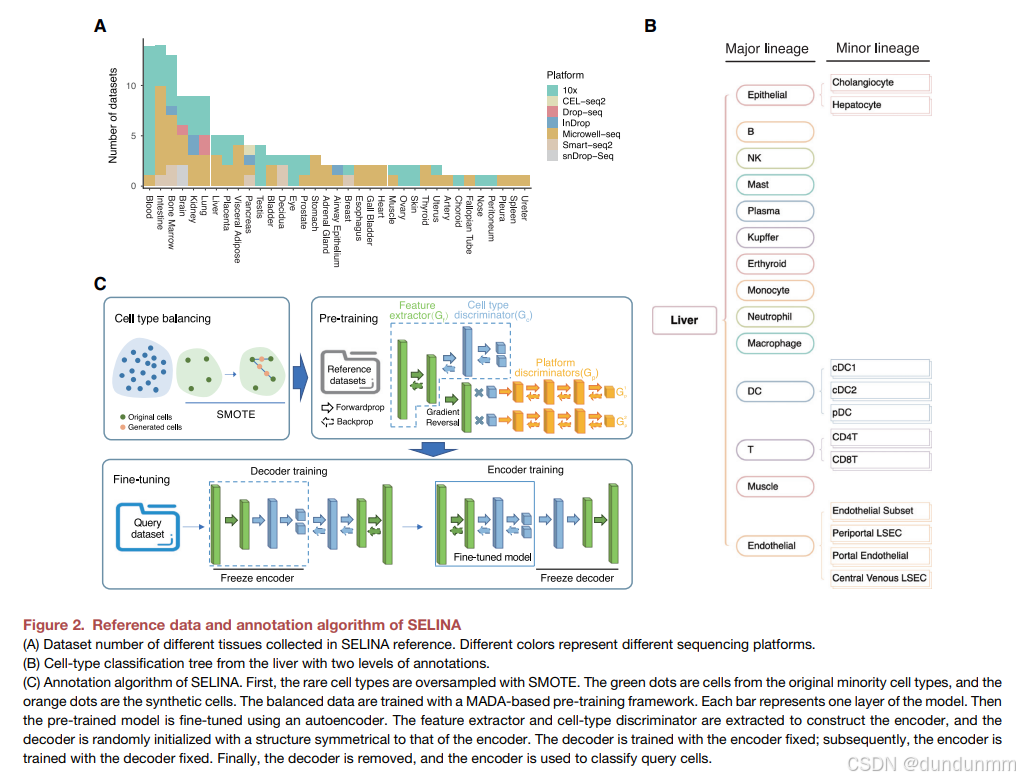
在每个组织中,不同数据集之间不一致的细胞类型名称被统一,随后根据文献和 Cell Ontology²⁹ 被划分为主类和次类。以肝脏为例,主干谱系包含 14 种不同的细胞类型,其中仅树突状细胞(DC)、内皮细胞和上皮细胞包含亚谱系(图 2B)。通过聚合所有组织,我们构建了一个全面的人类细胞类型本体树,用于描述 scRNA-seq 数据中细胞的父子关系。通过引入并整理已发表研究中的细胞类型,SELINA 为用户提供了更为统一的标准,以规范和组织 scRNA-seq 数据中的细胞类型图谱,并支持输入数据的注释。
SELINA 结合 SMOTE、MADA 与自编码器提升注释精度
SELINA 的注释算法包括三个步骤:细胞类型平衡、预训练和微调(图 2C)。在大多数分类算法中,分类器往往会为了获得更高的整体训练精度而错误分类少数类样本,代价是学习成本较低。尤其当少数类样本同时出现在训练集和测试集中时,分类器难以在测试中正确识别它们,因为训练样本仅能提供有限信息。
数据增强技术可以缓解数据不平衡问题,其中 SMOTE²³ 是一种经典算法。scRNA-seq 数据存在明显的类别不均衡:在我们的参考中,少数类细胞类型通常仅包含数十个细胞,而多数类细胞类型可以达到数万个。因此,SELINA 首先采用 SMOTE 对少数类细胞进行过采样。对于稀有细胞类型中的两两随机细胞,SELINA 将它们的基因表达向量分别乘以随机权重后相加,生成一个合成细胞。通俗地说,生成的细胞就是原始细胞的线性组合,随机落在这两个细胞连线的某个位置。该过程会持续进行,直到稀有细胞类型的数量达到与多数类细胞类型相同的量级,或不少于 1,000 个。
在 预训练阶段,SELINA 使用 MADA²⁴ 去除由不同测序平台引入的批次噪声。预训练框架由三部分组成:特征提取器、细胞类型判别器和测序平台判别器。特征提取器生成的特征向量会同时输入细胞类型判别器和平台判别器。与传统的对抗神经网络³¹不同,SELINA 的平台判别器包含多个分类器,其数量等于细胞类型的数量。对于某个特定平台分类器,输入特征向量会与该细胞被判定为对应细胞类型的概率相乘,该概率由细胞类型判别器计算。在平台预测误差的反向传播过程中,特征向量的梯度会被反转,使得特征提取器被训练为最大化平台判别器的损失,而平台判别器被训练为最小化损失。因此,随着训练进行,特征提取器生成的特征对于平台判别器来说越来越难以分类;即使输入特征之间的差异已非常微小,平台判别器依然会尽力区分平台来源,直到差异几乎完全消除。该策略能够通过单独捕捉每种细胞类型的批次信息,并在对抗方式下训练特征提取器与平台判别器,从而实现不同测序平台表达分布的细粒度对齐。
在 MADA 的原始应用场景中,域判别器主要用于帮助提取参考数据和查询数据之间的共性特征。而在 SELINA 中,MADA 被用于揭示参考数据中不同平台之间的共性信息。因此,参考数据与查询数据之间的差异仍然存在。为减少这种差异,SELINA 在 微调步骤中引入了自编码器。其中,编码器由预训练模型的特征提取器和细胞类型判别器构成,已经学习了来自大量参考数据的特征转换;而解码器由于参数随机初始化,仍需要调整。SELINA 首先冻结编码器,仅更新解码器参数,以保持预训练模型中学到的转换不被破坏。当总损失收敛后,SELINA 冻结解码器并更新编码器参数。经过编码器的再次训练,重构损失会进一步下降,从而使编码器能够根据查询数据的分布进行调整,最终减少参考数据与查询数据之间的批次噪声。

有“AI”的1024 = 2048,欢迎大家加入2048 AI社区
更多推荐
 10
10 0
0- 0



 已为社区贡献11条内容
已为社区贡献11条内容

所有评论(0)